Section SV4 Module de Biochimie Métabolique Partie - FSA

0
[ S ou P ] PPRS Phase Stationnaire PPOS
[P]
S1
dS
S2
P1
dP
P2 [S]
[ES]
t0t1t2Temps en min
dt
Section SV4
Module de Biochimie Métabolique
Partie Enzymologie
Année universitaire: 2014-2015
Pr Najat ALIF
dS dP
v = = = Cte
dt dt
d[ES]
= 0
dt

1
TABLE DES MATIERES
CINETIQUE DES REACTIONS CHIMIQUES.................................................................................1
1) Vitesse de réaction.......................................................................................................................1
1. 1) Vitesse initiale .....................................................................................................................1
1.2) Vitesse instantanée................................................................................................................1
2) Ordre d'une réaction.....................................................................................................................2
2. 1) Réaction d'ordre zéro...........................................................................................................2
2. 2) Réactions du premier ordre..................................................................................................2
2.3) Réactions du deuxième ordre................................................................................................4
2.4) Réversibilité..........................................................................................................................4
ASPECTS GENERAUX SUR LES ENZYMES.................................................................................5
1) Définitions ...................................................................................................................................5
1.1 Enzyme...................................................................................................................................5
1.2 Catalyseur...............................................................................................................................5
1.3 Substrat...................................................................................................................................5
1.4 Produit....................................................................................................................................5
1.5 Cofacteur................................................................................................................................5
1.6 Coenzyme et groupe prosthétique..........................................................................................5
1.7 Site actif: lieu de la catalyse...................................................................................................5
2) Propriétés générales des enzymes................................................................................................5
2.1) Catalyse.................................................................................................................................5
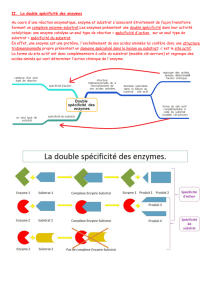
2.2) Spécificité.............................................................................................................................6
2.2.1) Spécificité de réaction....................................................................................................6
2.2.2) Spécificité du substrat....................................................................................................6
2.2.3) Stéréospécificité.............................................................................................................6
3. Classification des enzymes ..........................................................................................................6
CINETIQUE ENZYMATIQUE CLASSIQUE...................................................................................9
1) Le complexe enzyme-substrat :...................................................................................................9
1.1 Modèle de représentation selon Fischer.................................................................................9
1.2 Structure élastique des protéines: l'ajustement induit "induced fut"......................................9
2) Cinétique enzymatique fondamentale..........................................................................................9
2.1 Hypothèse de l'état stationnaire selon Michaelis-Menten......................................................9
2-1-1- Phases de la réaction.....................................................................................................9
2.1.2 Mesure de la vitesse de la réaction................................................................................10
2.1.3 Effet de la concentration d’enzyme ..............................................................................11
2.1.4 Effet de la concentration en substrat.............................................................................12
2.2 Cinétique Michaelienne .......................................................................................................13
2.3 Représentation graphique.....................................................................................................15
3) Effecteurs réversibles de la réaction enzymatique.....................................................................16

2
3.1 Effecteur...............................................................................................................................16
4. Inhibitions enzymatiques ...........................................................................................................16
4.1 Inhibition compétitive..........................................................................................................16
4.1.1 Cinétique de l’inhibition compétitive ...........................................................................17
4.1.2 Exemples d’inhibition compétitive...............................................................................18
4.1.3 Représentation graphique en hyperbole........................................................................18
4.2 Inhibition non compétitive..................................................................................................18
4.2.1 Cinétique de l’inhibition non compétitive ....................................................................19
4.2.2 Exemples d’inhibitions non compétitives.....................................................................20
4.2.3 Représentations graphiques...........................................................................................21
4.3 Inhibition anti (ou un) compétitive (équation).....................................................................21
4.3.1 Cinétique de l’inhibition incompétitive ........................................................................21
4.3.2 Représentations graphiques...........................................................................................22
4.4 Cas particulier : inhibition par excès de substrat .................................................................23
4.5 Rôles biologique de l’inhibition...........................................................................................23
4.6 Tableau (10) comparatif des paramètres cinétiques pour différents types d’inhibition.......24
4.7. Tableau (11) Tableau comparatif des différentes représentations cinétiques pour différents
types d’inhibitions......................................................................................................................25
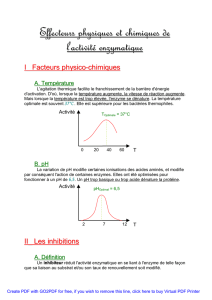
5) Influence du pH sur l'activité enzymatique ...........................................................................26

1
CINETIQUE DES REACTIONS CHIMIQUES
1) Vitesse de réaction
La vitesse d'une réaction chimique est définie par la quantité de substances transformées ou
de produit formé par unité de temps. Les facteurs qui influencent la vitesse de la réaction sont: la
concentration, la température et la présence de catalyseurs.
1. 1) Vitesse initiale A + B → C + D
A et B sont les substances réagissantes et C et D sont les produits.
vm=C/t = (C1- C0)/(t1- t0) vm= - A/t = - (A1- A0)/(t1- t0)
La vitesse initiale est mesurée par la pente de la tangente à la courbe au temps 0.
1.2) Vitesse instantanée
La vitesse de la réaction varie le plus souvent entre son début et sa fin. A chaque
temps, on peut définir une vitesse instantanée qui est égale à la pente de la tangente à la courbe de
vitesse au point correspondant à ce temps.
v= dC/dt = (C2- C1)/(t2- t1) v= - dA/dt = - (A2- A1)/(t2- t1)

2
2) Ordre d'une réaction
A + B → C + D
v = k [A]m[B]nk= Constante de vitesse.
La vitesse d'une réaction est fonction des concentrations des substances réagissantes.
La somme m + n est appelé ordre de la réaction. C'est une valeur expérimentale obtenue à partir
d'une série de détermination de la vitesse en fonction des concentration des substances réagissante.
Elle permet de caractériser le mécanisme d'une réaction. Elles est distincte de la stœchiométrie (on
ne doit pas la confondre avec le nombre de molécules réagissantes.
2. 1) Réaction d'ordre zéro
Quand m + n = 0, on dit que la réaction est d'ordre zéro.
d[A]
v= - ———— =ko, La vitesse est constante.
dt
La quantité de produit formé est linéaire en fonction du temps.
Ce type de réaction , dite d'ordre 0, s'observe en milieu hétérogène, par exemple, lorsque la
substance réagissante, peu soluble, est présente en quantité supérieure à sa solubilité. Cet excès se
dissout à mesure que la réaction progresse et maintient la saturation de la solution. L'ordre de la
réaction cesse d'être nul lorsque la concentration devient inférieur à la saturation.
Une cinétique d'ordre nul s'obtient également lorsque la substance dont on mesure la
variation résulte d'une étape intermédiaire de la réaction avec participation d'une substance
étrangère, comme le complexe enzyme-substrat pendant la phase stationnaire des réactions
enzymatiques.
2. 2) Réactions du premier ordre
Quand m + n = 1, on dit que la réaction est de premier ordre. C'est le cas d'une molécule qui
se transforme en une autre. Par exemple une protéine qui se dénature, un radioélément qui se
désintègre, mutarotation de sucres, isomérisation, racémisation d'acides aminés...
Considérons la réaction de transformation de A en B : A B
Au cours de la réaction la vitesse de la réaction varie et ne peut être considérée comme
constante que pendant un intervalle de temps infiniment petit. Le nombre de molécules de produit B
qui apparaissent pendant cet intervalle est évidement égal au nombre de molécules A qui
disparaissent. On a donc : d[A] d[B]
v = = = k1A
dt dt
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
1
/
32
100%