Le complexe de régulation de l`homéostasie du Cholestérol

1
Logez Christel
Delbos Lila
Le complexe de
régulation de
l’homéostasie du
Cholestérol
Master EGPR 7

2
Sommaire
RESUME...................................................................................................................................3
LISTE DES ABREVIATIONS ...............................................................................................4
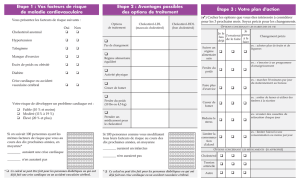
I. INTRODUCTION :..............................................................................................................5
1) Bref historique de la découverte du système de régulation de l’homéostasie du
cholestérol : ............................................................................................................................ 6
2) Présentation des protéines impliquées dans le complexe : ................................................7
3) Mécanisme du complexe de régulation du Cholestérol :...................................................8
II. RESULTATS :.....................................................................................................................9
1) Le 25-HC et le Cholestérol induisent tous deux la formation du complexe SCAP/Insig..9
2) Le 25-HC n’induit pas de changement de conformation de SCAP et l’inhibition du
clivage de SREBP est dépendante de Insig-1.......................................................................10
3) Le 25-HC n’interagit pas directement avec SCAP :........................................................11
4) Découverte d’une nouvelle protéine interagissant avec les protéines SCAP et Insig :
PGRMC1 :............................................................................................................................12
III. PROJET DE RECHERCHE :........................................................................................14
1) Présentation de PGRMC1 :..............................................................................................14
2) Modèle d’étude ................................................................................................................15
3) PGRMC1 intervient-elle dans l’inhibition du clivage de SREBP ? ................................15
A. Transfection stable des cellules COS7 :......................................................................16
B. Extinction de PGRMC1 :............................................................................................. 16
C. Expérience :.................................................................................................................16
4) Le 25-HC interagit-il directement avec PGRMC1 ?........................................................18
A. Production du photo-25-HC radioactif :.....................................................................18
B. Expérience :.................................................................................................................18
5) Y a t-il formation d’un complexe multiprotéique PGRMC1/SCAP/Insig ?....................19
IV. CONCLUSIONS :............................................................................................................21
RÉFÉRENCES :.....................................................................................................................23
ANNEXE.................................................................................................................................25

3
Résumé
La concentration en cholestérol, constituant majeur des membranes lipidiques et
précurseur de nombreuses hormones stéroïdiennes doit être régulée car une trop forte
concentration en cholestérol peut conduire à la mort cellulaire ou à une athérosclérose. La
synthèse de cholestérol est autorégulée grâce à un contrôle en feedback faisant intervenir un
complexe constitué des protéines SREBP, SCAP et Insig. La fixation du cholestérol sur
SCAP induit la formation d’un complexe SREBP/SCAP/Insig retenu dans la membrane du
RE qui empêche SREBP de jouer son rôle d’activateur de la transcription des gènes impliqués
dans la synthèse du cholestérol en inhibant son clivage.
Le 25-Hydroxycholestérol peut également inhiber le clivage de SREBP en induisant la
formation du complexe SCAP/Insig mais sans interagir directement avec SCAP. Ceci suggère
qu’une protéine non identifiée activée par le 25-HC induirait la formation du complexe
SCAP/Insig pour inhiber le clivage de SREBP. Une récente étude, qui a mis en évidence que
la protéine PGRMC1 interagit directement avec SCAP et Insig, soulève l’hypothèse selon
laquelle PGRMC1 pourrait être cette protéine. Cependant, à ce jour, aucune étude n’a été
réalisée pour déterminer le rôle de PGRMC1 dans le mécanisme de régulation de
l’homéostasie du cholestérol par la voie du 25-HC. C’est pourquoi nous nous sommes
intéressées à cette protéine et avons élaboré un projet de recherche, faisant intervenir des
expériences de photo-pontage et de siRNA, dans le but de vérifier l’intervention de PGRMC1
et de comprendre son rôle potentiel dans ce mécanisme de régulation.

4
Liste des abréviations
25-HC : 25-Hydroxycholestérol
HA : Hémaglutinine
HSV : Herpès Simplex Virus
INSIG : Insulin Induced Gene
PGRMC1 : Progesterone Receptor Membrane Component 1
RE : Réticulum Endoplasmique
S1P : Sérine Protéase 1
S2P : Sérine Protéase 2
SCAP : SREBP Cleavage Activating Protein
SDS : Sodium Dodecyl Sufate
PAGE : Poly Acrylamide Gel Electrophoresis
SRE : Sterol Regulatory Element
SREBP : Sterol Regulatory Element Binding Protein

5
Figure1 :
Voie de biosynthèse du cholestérol.
Enzymes, substrats produits, inhibiteurs des
enzymes.
(www.answers.com/topic/cholesterol)
I. Introduction :
Le cholestérol est une molécule essentielle chez les eucaryotes supérieurs. En effet il est
l’un des constituants majeurs des membranes lipidiques et a pour fonction de diminuer la
perméabilité membranaire et d’augmenter la fluidité membranaire. De plus, le cholestérol est
le précurseur métabolique des hormones stéroïdiennes comme la progestérone et dans le foie,
le cholestérol est le précurseur de l’acide glycocholique, un composant majeur de la bile. Les
oxystérols, issus du métabolisme du cholestérol, interviennent également en tant que
régulateurs de nombreuses voies métaboliques. Les besoins quotidiens en cholestérol d’un
adultes sont d’environ un gramme et demi, et le corps trouve cet approvisionnement pour un
tiers dans l’alimentation et les deux tiers restants sont synthétisés dans le foie.
Cependant il est nécessaire pour les cellules de réguler leur concentration en cholestérol
afin de maintenir un taux physiologique n’excédant pas deux grammes de cholestérol par
millilitre de sang. En effet une trop forte concentration en cholestérol peut détruire les
fonctions des membranes cellulaires et l’accumulation de cholestérol peut conduire à la mort
cellulaire ou à une athérosclérose.
De ce fait, au cours de l’évolution les eucaryotes supérieurs ont acquis un mécanisme
qui permet de réguler cette concentration.
Ce mécanisme fait intervenir un système
de régulation en feedback qui contrôle le
niveau de cholestérol dans les membranes
cellulaires et module la transcription des
gènes codant les enzymes HMGCoA
synthase, la HMGCoA réductase,… . En
effet, ces enzymes interviennent dans la
biosynthèse du cholestérol à partir de
l’acétyl-CoA au niveau du cytosol pour la
HMG-CoA synthase et au niveau du RE
pour la HMG-CoA réductase. (figure1).
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
1
/
25
100%