Dossier L Antagonistes des hormones peptidiques Les antagonistes de la GnRH

Antagonistes des hormones peptidiques
Dossier
Les antagonistes de la GnRH
N. Chabbert-Buffet*, P. Bouchard**
L
a découverte du rôle crucial
de la GnRH dans la vie repro-
ductive a été rapidement suivie
par la mise au point de traitements de
l’infertilité faisant appel à la GnRH
elle-même ou à des analogues de la
GnRH. La GnRH native administrée
de manière pulsatile à l’aide de mini-
pompes (1) a démontré son effica-
cité dans le traitement de l’infertilité
d’origine hypothalamique. Les pre-
miers analogues de la GnRH ont été
synthétisés dans les mois qui ont
suivi la découverte de la structure
décapeptidique de la GnRH en 1971
par l’équipe de Schally (2). Parmi
les analogues de la GnRH, on dis-
tinguait les agonistes et les antago-
nistes de la GnRH. Dès 1978, il fut
clair que l’administration répétée
d’agonistes de la GnRH aboutissait
en fait à une inhibition de la fonction
gonadotrope avec un effondrement
de la sécrétion des stéroïdes sexuels,
cela après une phase initiale de sti-
mulation (3). Plusieurs années furent
nécessaires pour élucider ce phéno-
mène. La mise au point de dosages
spécifiques et sensibles de la LH a
permis de démontrer que, parallèle-
ment à la chute des stéroïdes (estra-
diol ou testostérone), les niveaux de
LH étaient bas. Enfin, il a été montré
que ce phénomène était dû à la
désensibilisation des récepteurs de
la GnRH au niveau de la membrane
des cellules gonadotropes hypo-
physaires, comparable à ce que l’on
observe en cas d’administration
continue (et non pulsatile comme en
physiologie) de GnRH (4, 5).
Les antagonistes de la GnRH ont été
découverts dès 1972, et leur méca-
nisme d’action est complètement
différent. Ils se lient au récepteur
de la GnRH avec une haute affinité
mais sont incapables d’activer la cas-
cade post-récepteur de transduction
du signal. lls se comportent donc
comme des inhibiteurs compétitifs
du récepteur (6-8).
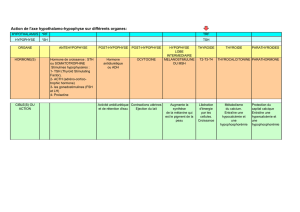
Structure et mécanisme
d’action des antagonistes
de la GnRH (figure 1, tableau I)
Structure
Les antagonistes de la GnRH sont pro-
duits par substitution de 4 à 6 acides
aminés de la chaîne décapeptidique
native de GnRH (figure 1). Ceux-
ci sont remplacés par des acides
aminés dextrogyres non physio-
logiques. Le premier antagoniste a été
obtenu par la suppression de l’histi-
dine en position 2, dont le rôle dans
* Service de médecine interne, hôpital Tenon, et
EA 1533 Génétique de la reproduction humaine,
CHU Saint-Antoine, Paris.
** Service d’endocrinologie et EA 1533 Génétique
de la reproduction humaine, CHU Saint-Antoine,
Paris.
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
31
Figure 1. Mécanisme d’action des antagonistes de la GnRH par opposition aux agonistes.

l’interaction avec le récepteur est
majeur. L’utilisation clinique des
antagonistes de la GnRH a été long-
temps limitée par les phénomènes
d’intolérance cutanée ou systé-
mique, observés avec les composés
de première génération du fait de
leur capacité histaminolibératrice.
Cela concernait en particulier les
molécules comportant une arginine
dextrogyre en position 6 (DArg6).
Une dizaine de nouvelles molécules
quasi dépourvues d’effet allergisant
sont actuellement en cours d’étude
clinique (9, 10) et deux molécules
sont commercialisées (Cetrorelix®
et Ganirelix®,tableau II) (11, 12).
Pharmacologie
L’administration sous-cutanée d’an-
tagonistes de la GnRH produit une
chute rapide des taux de LH et de
stéroïdes sexuels, suivie par une
décroissance plus lente de la FSH.
Celle-ci n’est significative qu’après
plusieurs injections d’antagonistes
(13). Cette décroissance de la LH et
de la FSH sous antagonistes de la
GnRH vient confirmer qu’il n’existe
qu’une seule “gonadolibérine” hypo-
thalamique, commune à LH et FSH,
et justifie le remplacement du terme
LHRH par GnRH.
L’activité biologique des antago-
nistes de la GnRH nécessite l’admi-
nistration de milligrammes de ces
molécules. Les agonistes de la GnRH,
en revanche, sont actifs à des doses de
quelques dizaines de microgrammes.
L’une des explications proposées est
que les antagonistes doivent occuper
tous les sites récepteurs en perma-
nence, alors que les agonistes – pro-
voquant une désensibilisation, et
donc une diminution des sites récep-
teurs – sont actifs à des doses plus
faibles. Cependant, certains auteurs
ont décrit in vitro une diminution des
sites récepteurs sous antagonistes
(14). Ces données semblent ne pas
être extrapolables in vivo, car l’ad-
ministration d’agonistes de la GnRH
à des patients sous antagonistes
induit une élévation transitoire des
gonadotrophines (15). Cela suggère
donc que les récepteurs sont présents
sur la membrane.
Les propriétés pharmacocinétiques
des antagonistes sont complexes, leur
demi-vie pouvant varier de quelques
heures à plus de trente heures en
fonction de la dose administrée. Les
antagonistes ont également la pro-
priété de former un gel qui leur
confère une libération prolongée. La
formation du gel survient probable-
ment au site d’injection. In vitro, elle
dépend des concentrations de NaCl.
Cette propriété a été utilisée pour
produire des antagonistes d’action
prolongée. Les données cliniques au
long cours manquent cependant.
Le composé oral TAK 103, premier
antagoniste non peptidique, semble
avoir une longue durée d’action pro-
longée (16).
Les agonistes induisent initialement
une élévation des gonadotrophines
et des stéroïdes sexuels. C’est l’effet
flare up,qui peut être indésirable
lorsque les agonistes sont utilisés
pour traiter une maladie hormono-
dépendante (cancer de la prostate ou
du sein, puberté précoce centrale).
Au contraire, cet effet peut être, par
exemple, utilisé pour déclencher le
pic ovulatoire de LH au cours des
cycles de fécondation in vitro (FIV).
Secondairement, le phénomène de
désensibilisation qui empêche le recy-
clage des récepteurs à la membrane
entraîne une inhibition gonadotrope
et une chute des stéroïdes sexuels.
Les antagonistes ont un effet d’em-
blée inhibiteur, dont la durée est liée
à la pharmacocinétique du composé
utilisé et à la dose. L’effet apparaît
en quelques heures et dure de 10 à
100 heures. L’effet suppresseur des
antagonistes sur le niveau de gonado-
trophines est également plus marqué
que celui des agonistes (13). Ils sup-
priment de manière aussi efficace
LH et FSH, alors que les agonistes
ont un effet sur la LH sans affecter
la sécrétion de FSH, qui augmente
même avec le temps. Ils seraient
donc a priori plus intéressants dans
le traitement des affections hormo-
nodépendantes, mais leur durée
d’action cliniquement limitée les
rend inadéquats dans ce contexte.
Dossier
Dossier
32
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
Abarelix Ac-DNal-DCpa-DPal-Ser-N-Me Tyr-Dasn-Leu-Ilys-Pro-DAla- NH2
Acyline Ac-DNal-DCpa-DPal-Ser-Aph-Daph(Ac)-Leu-ILys-Pro-DAla- NH2
Antarelix Ac-DNal-DCpa-DPal-Ser-Tyr-DHci-Leu-ILys-Pro-DAla-NH2
Iturelix Ac-DNal-DCpa-DPal-Ser-Lys(Nic)-Dlys(Nic)-Leu-Ilys-Pro-DAla- NH2
Azaline B Ac-DNal-DCpa-DPal-Ser-Aph(Atz)-DAph(Atz)-Leu-Ilys-Pro-DAla- NH2
Nal-Glu Ac-DNal-DCpa-DPal-Ser-Arg-Dglu(AA)-Leu-Arg- Pro-DAla- NH2
FE 200486 Ac-DNal-DCpa-DPal-Ser-Aph(Hor)-DAph(Cba)-Leu-ILys-Pro-DAla- NH2
Cetrorelix** Ac-DNal-DCpa-DPAla-Ser-Tyr-Dcit-Leu-Arg-Pro-DAla- NH2
Ganirelix*** Ac-DNal-DCpa-DPal-Ser-Tyr-DHArg(Et2)-Leu-HArg(Et2)-Pro-DAla- NH2
GnRH pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly- NH2
Tableau I. Structure des antagonistes de la GnRH.
Nom
®
(DCI) Laboratoire Voie Dosages Coût par cycle
Cétrotide
®
Serono sous-cutané 0,25 et 3 mg 270 à 380 euros
(Cétrorélix) env.
Orgalutran
®
Organon sous-cutané 0,25 mg 240 à 330 euros
(Ganirelix) env.
Tableau II.Antagonistes de la GnRH commercialisés en France.

Dossier
Dossier
Le développement de formes orales de
longue durée d’action pourra peut-
être apporter une solution à l’avenir.
Mécanisme d’action
aux niveaux cellulaire
et moléculaire
Les analogues de la GnRH inter-
agissent avec le récepteur de type 1
de la GnRH, auquel ils se lient avec
une haute affinité. Le récepteur de
la GnRH est une molécule à sept
domaines transmembranaires, cou-
plée à une protéine G (Gq11) qui
assure la transmission intracellulaire
du signal. Le domaine extracellulaire
de grande taille est le site de liaison
du ligand et vient présenter ce der-
nier aux sept domaines transmem-
branaires organisés en puits. Cela
permet de déclencher la cascade de
transduction du signal. Il semble
également que les récepteurs de
la GnRH puissent se dimériser et
coopérer après liaison d’un agoniste.
Les antagonistes se lient au récepteur
mais n’induisent aucune activation
de la transduction du signal. Les
effets des antagonistes sur les phéno-
mènes de dimérisation du récepteur
de la GnRH sont encore mal connus,
mais il semble que la dimérisation
n’ait pas lieu (17). Enfin, le site précis
de liaison des agonistes et des anta-
gonistes de la GnRH au récepteur
semble différer (18).
Applications cliniques
Étant donné les caractéristiques des
antagonistes, il est logique d’envi-
sager leur utilisation dans des situa-
tions où une suppression rapide et
profonde de la sécrétion des gonado-
trophines est nécessaire. C’est le cas
au cours des cycles de procréation
médicalement assistée.
Chez les femmes normales, le pic
préovulatoire de LH est déclenché
par l’action permissive hypothala-
mique de l’estradiol, sécrété à des
taux de 150 à 200 pg/ml par le folli-
cule dominant (19). L’administration
d’un antagoniste de la GnRH est
capable de retarder le pic de LH si
elle a lieu en fin de phase follicu-
laire, avant le pic de LH ou même
au moment de celui-ci (20-22). Il est
bien sûr impossible de déterminer le
taux de GnRH portal dans l’espèce
humaine, mais il a été montré chez
le rat, les ovins et les primates que
la GnRH jouait un rôle crucial dans
le déclenchement du pic de LH (23-
27). La quantité de GnRH néces-
saire pour induire le pic de LH reste
inconnue. Chez la femme, il semble
que la GnRH ait un effet permissif
et qu’une augmentation de la GnRH
ne soit pas nécessaire pour induire
le pic de LH (28).
Au cours de l’hyperstimulation ova-
rienne contrôlée réalisée en FIV, les
taux d’estradiol, très importants en
raison du développement multifolli-
culaire, peuvent induire un pic de
LH prématuré. Ces pics de LH pré-
maturés entraînent une lutéinisation
prématurée du follicule et sont asso-
ciés à un taux d’échec de grossesse
plus important (29). Ils entraînent
donc l’annulation du cycle en cours.
Les antagonistes de la GnRH per-
mettent de retarder le pic de LH.
Les agonistes de la GnRH sont
également utilisés en FIV pour
“annuler” la fonction gonadotrope
endogène de la femme et prévenir
toute possibilité de pic prématuré de
LH. Ils sont pour cela utilisés selon
différents protocoles induisant une
désensibilisation plus ou moins pro-
longée. Ils permettent également une
meilleure organisation des plannings
de déclenchement de l’ovulation
des patientes. Les antagonistes de la
GnRH, disponibles de manière plus
récente, sont en cours d’évaluation
par rapport à ces traitements de réfé-
rence. Les antagonistes sont capables
de réduire la durée de stimulation
ovarienne et donc le nombre d’am-
poules de FSH utilisées. Ils rédui-
sent également le nombre d’hyper-
stimulations ovariennes. Toutefois,
ils entraînent également une baisse
significative, bien que modérée, des
taux de grossesse par rapport aux
cycles traités par agonistes (30). Cela
nécessitera pour l’avenir une adap-
tation des protocoles de traitement.
Les protocoles actuellement utilisés
sont de deux types : mono-injection
de 3 mg au 7ejour de la stimulation
par FSH (sous forme d’HMG qui
comporte de la LH en faible quan-
tité et de la FSH extractives ou sous
forme recombinante pure), ou multi-
injection de 0,25 mg/jour du 6eau
13ejour de la stimulation environ.
Au cours des cycles de FIV sous
agonistes, la désensibilisation des
récepteurs contraint à déclencher
l’ovulation par de l’hCG, qui mime
l’action de la LH. Toutefois, la durée
de vie de l’hCG est importante, et
l’injection peut entretenir, voire
aggraver, une hyperstimulation pré-
existante. Au cours des cycles de FIV
sous antagonistes, on peut utiliser
l’effet flare up des agonistes (31)
pour déclencher l’ovulation. Une
injection d’agoniste d’action brève
permet de déclencher un pic de LH
endogène, a priori plus physiologique
que l’injection d’hCG.
Après l’ovulation, le développement
du corps jaune et sa survie sont
dépendants de la LH. Lors des cycles
de FIV traités par agonistes de la
GnRH, l’inhibition gonadotrope est
prolongée et impose un soutien de
la phase lutéale, par injection d’hCG
(avec les risques d’hyperstimulation
déjà évoqués) ou par administration
de progestérone. Lors des cycles sous
antagonistes, il semble que, malgré
la durée d’action brève de ces com-
posés, la phase lutéale soit raccourcie
et nécessite une supplémentation
progestative (32-33).
Les effets des antagonistes de la
GnRH sur l’enfant à venir sont encore
mal connus mais semblent limités ou
nuls (34, 35). Les embryons obtenus
après un cycle sous antagonistes
semblent normaux selon les critères
morphologiques “grossiers” dont nos
disposons actuellement. Ils permet-
tent d’obtenir après congélation un
taux de grossesse évolutive compa-
rable à celui que l’on observe avec les
embryons congelés issus de cycles
sous agonistes de la GnRH (36). De
manière plus générale, les taux de
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
33

malformation fœtale au cours des
grossesses obtenues après traitement
par antagonistes ne semblent pas
plus élevés que pour les autres tech-
niques de procréation médicalement
assistée (34, 35).
Conclusions
et perspectives
Les antagonistes de la GnRH repré-
sentent un outil potentiellement
formidable sur lequel il nous reste
beaucoup à découvrir. Les méca-
nismes d’action sont encore mal
connus, et la recherche dans ce
domaine contribue à la fois à élucider
ces mécanismes et à mieux connaître
la pharmacologie moléculaire des
récepteurs de la GnRH (37). Les
protocoles d’utilisation dans le cadre
de la FIV doivent également être
améliorés (38). Le développement
de molécules de longue durée d’ac-
tion pourra peut-être permettre
d’élargir les indications des antago-
nistes de la GnRH aux pathologies
hormonodépendantes, comme le
cancer du sein ou de la prostate et la
puberté précoce centrale. D’autres
indications à plus court terme sont
en cours d’évaluation, comme la pré-
paration à l’hystéroscopie en cas de
saignement, à la mammographie en
cas de mastodynies très importantes
ou de densité mammaire excessive.
Dans le traitement des cancers hor-
monodépendants, il n’existe aucune
raison de penser que les antagonistes
seront supérieurs aux agonistes, qui
suppriment de manière très efficace
les taux de stéroïdes sexuels avec
des résultats cliniques identiques à
ceux obtenus après castration. De
plus, les formulations “longue durée”,
indispensables au confort des patients,
n’existent pour le moment que pour
les agonistes de la GnRH. Enfin, la
mise au point de formes orales
actuellement en cours est une étape
majeure du développement de ces
molécules (16, 39).
Références
1.
Leyendecker G, Wildt L, Hansmann M. Pre-
gnancies following chronic intermittent (pulsatile)
administration of Gn-RH by means of a portable
pump (“Zyklomat”) : a new approach to the treat-
ment of infertility in hypothalamic amenorrhea.
J Clin Endocrinol Metab 1980 ; 51: 1214-6.
2.
Baba Y, Matsuo H, Schally AV. Structure of the
porcine LH- and FSH-releasing hormone. II.
Confirmation of the proposed structure by conven-
tional sequential analyses. Biochem Biophys Res
Commun 1971 ; 44: 459-63.
3.
Cusan L, Auclair C, Belanger A et al. Inhibitory
effects of long term treatment with a luteinizing
hormone-releasing hormone agonist on the pitui-
tary-gonadal axis in male and female rats. Endo-
crinology 1979 ; 104 : 1369-76.
4.
Casper RF. Clinical uses of gonadotropin-
releasing hormone analogues. CMAJ 1991 ; 144 :
153-8.
5.
Knobil E. The neuroendocrine control of the
menstrual cycle. Recent Prog Horm Res 1980;
36 : 53-88.
6.
Karten MJ, Rivier JE. Gonadotropin-releasing
hormone analog design. Structure-function studies
toward the development of agonists and antago-
nists : rationale and perspective. Endocr Rev
1986 ; 7: 44-66.
7.
Bouchard P, Garcia E. Comparison of the me-
chanisms of action of LHRH analogs and steroids
in the treatment of endometriosis. Contrib Gyne-
col Obstet 1987 ; 16: 260-5.
8.
Bouchard P, Wolf JP, Hajri S. Inhibition of ovu-
lation : comparison between the mechanism of
action of steroids and GnRH analogues. Hum Re-
prod 1988 ; 3: 503-6.
9.
Felberbaum R, Ludwig M, Diedrich K. Agonists
and antagonists, formulation and indication in
GnRH analogues. Philadelphia : WB Sauders
Company, 2001.
10.
Blithe DL. Applications for GnRH antagonists.
Trends Endocrinol Metab 2001 ; 12 : 238-40.
11.
Gillies P, Faulds D, Balfour J, Perry C. Ga-
nirelix. Drugs 2000 ; 59 : 107-111.
12.
Reissmann T, Schally AV, Bouchard P et al.
The LHRH antagonist cetrorelix : a review. Hum
Reprod Update 2000 ; 6: 322-31.
13.
Pavlou SN, Brewer K, Farley MG et al. Com-
bined administration of a gonadotropin-releasing
hormone antagonist and testosterone in men in-
duces reversible azoospermia without loss of li-
bido. J Clin Endocrinol Metab 1991 ; 73 : 1360-9.
14.
Kovacs M, Schally AV, Csernus B, Rekasi Z.
Luteinizing hormone-releasing hormone (LH-RH)
antagonist Cetrorelix down-regulates the mRNA
expression of pituitary receptors for LH-RH by
counteracting the stimulatory effect of endoge-
nous LH-RH. Proc Natl Acad Sci USA 2001 ; 98 :
1829-34.
15.
Lahlou N, Delivet S, Bardin CW et al. Changes
in gonadotropin and alpha-subunit secretion after
a single administration of gonadotropin-releasing
hormone antagonist in adult males. Fertil Steril
1990 ; 53: 898-905.
16.
Suzuki N, Cho N, Furuya S et al. A novel, potent
and orally active nonpeptide antagonist for the
human gonadotropin-releasing hormone receptor.
The Endocrine Society 84th Annual meeting,
2002 : OR7-2.
17.
Conn PM, Crowley WF Jr. Gonadotropin-
releasing hormone and its analogs. Annu Rev Med
1994 ; 45: 391-405.
18.
Zhou W, Rodic V, Kitanovic S et al. A locus of
the gonadotropin-releasing hormone receptor that
differentiates agonist and antagonist binding sites.
J Biol Chem 1995 ; 270: 18853-7.
19.
Clarke IJ. Evidence that the switch from ne-
gative to positive feedback at the level of the pi-
tuitary gland is an important timing event for the
onset of the preovulatory surge in LH in the ewe.
J Endocrinol 1995 ; 145: 271-82.
20.
Ditkoff EC, Cassidenti DL, Paulson RJ et al.
The gonadotropin-releasing hormone antagonist
(Nal-Glu) acutely blocks the luteinizing hormone
surge but allows for resumption of folliculogenesis
in normal women. Am J Obstet Gynecol 1991 ;
165 : 1811-7.
21.
Christin-Maitre S, Olivennes F, Dubourdieu S
et al. Effect of gonadotrophin-releasing hormone
(GnRH) antagonist during the LH surge in normal
women and during controlled ovarian hyperstimu-
lation. Clin Endocrinol (Oxf) 2000 ; 52 : 721-6.
22.
Leroy I, d’Acremont M, Brailly-Tabard S et al.
A single injection of a gonadotropin-releasing
hormone (GnRH) antagonist (Cetrorelix) post-
pones the luteinizing hormone (LH) surge : further
evidence for the role of GnRH during the LH
surge. Fertil Steril 1994 ; 62 : 461-7.
23.
Moenter SM, Caraty A, Karsch FJ. The estradiol-
induced surge of gonadotropin-releasing hormone
in the ewe. Endocrinology 1990 ; 127: 1375-84.
24.
Xia L,Van Vugt D, Alston EJ et al. A surge of
gonadotropin-releasing hormone accompanies the
estradiol-induced gonadotropin surge in the rhesus
monkey. Endocrinology 1992 ; 131 : 2812-20.
25.
Norman RL, Rivier J, Vale W, Spies HG.
Inhibition of estradiol-induced gonadotropin
release in ovariectomized rhesus macaques by a
gonadotropin-releasing hormone antagonist. Fertil
Steril 1986 ; 45: 288-91.
26.
Clarke IJ. Variable patterns of gonadotropin-
releasing hormone secretion during the estrogen-
induced luteinizing hormone surge in ovariecto-
mized ewes. Endocrinology 1993 ; 133 : 1624-32.
27.
Karsch FJ, Bowen JM, Caraty A et al. Gonado-
tropin-releasing hormone requirements for ovu-
lation. Biol Reprod 1997 ; 56: 303-9.
28.
Martin KA,Welt CK, Taylor AE et al. Is GnRH
reduced at the midcycle surge in the human ? Evi-
dence from a GnRH-deficient model. Neuroendo-
crinology 1998 ; 67 : 363-9.
29.
Diedrich K, Ludwig M, Felberbaum RE. The
role of gonadotropin-releasing hormone antago-
nists in in vitro fertilization. Semin Reprod Med
2001 ; 19, 213-20.
30.
Al-Inany H, Aboulghar M. GnRH antagonist
in assisted reproduction : a Cochrane review. Hum
Reprod 2002 ; 17: 874-85.
31.
Olivennes F, Fanchin R, Bouchard P et al.
Triggering of ovulation by a gonadotropin-releasing
hormone (GnRH) agonist in patients pretreated
with a GnRH antagonist. Fertil Steril 1996 ; 66 :
151-3.
32.
Ragni G,Vegetti W, Baroni E et al. Comparison
of luteal phase profile in gonadotrophin stimulated
cycles with or without a gonadotrophin-releasing
hormone antagonist. Hum Reprod 2001 ; 16 :
2258-62.
Dossier
Dossier
34
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003

Dossier
Dossier
33.
Williams SC, Oehninger S, Gibbons WE et al.
Delaying the initiation of progesterone supple-
mentation results in decreased pregnancy rates
after in vitro fertilization : a randomized, pros-
pective study. Fertil Steril 2001 ; 76 : 1140-3.
34.
Ludwig M, Riethmuller-Winzen H, Felber-
baum RE et al. Health of 227 children born after
controlled ovarian stimulation for in vitro fertili-
zation using the luteinizing hormone-releasing
hormone antagonist cetrorelix. Fertil Steril 2001 ;
75 : 18-22.
35.
Olivennes F, Mannaerts B, Struijs M et al.
Perinatal outcome of pregnancy after GnRH anta-
gonist (ganirelix) treatment during ovarian stimu-
lation for conventional IVF or ICSI : a preliminary
report. Hum Reprod 2001 ; 16: 1588-91.
36.
Kol S. Embryo implantation and GnRH anta-
gonists : GnRH antagonists in ART: lower embryo
implantation ? Hum Reprod 2000 ; 15, 1881-2.
37.
Ott TR,Troskie BE, Roeske RW et al. Two mu-
tations in extracellular loop 2 of the human GnRH
receptor convert an antagonist to an agonist. Mol
Endocrinol 2002 ; 16: 1079-88.
38.
Fauser BC, Bouchard P, Coelingh Bennink H
et al. Alternative approaches in IVF. Human Re-
production Update 2002 ; 8: 1-9.
39.
Sasaki S, Cho N, Nara Y et al. Discovery of a
thieno[2,3-d]pyrimidine-2,4-dione bearing a p-
methoxyureidophenyl moiety at the 6-position : a
highly potent and orally bioavailable non-peptide
antagonist for the human luteinizing hormone-
releasing hormone receptor. J Med Chem 2003 ;
46 : 113-24.
Métabolismes Hormones Diabètes et Nutrition (VII), no1, janvier/février 2003
35
5
es
Entretiens de nutrition de l’Institut Pasteur de Lille
12 et 13 juin 2003
Au programme :– stress et nutrition (12 juin)
– l’allaitement maternel (13 juin)
Lieu :Institut Pasteur de Lille
Organisation scientifique :Dr Jean-Michel Lecerf
Contact :Marie-Françoise Tahon
Tél. : 03 20 87 71 88 – Fax: 03 20 87 72 96
E-mail : Marie-Francoise.Tahon@pasteur-lille.fr
Agenda
Agenda
1
/
5
100%