L L’hepcidine, l’hormone du fer Hepcidin, the iron hormone

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 4 - juillet-août 2009
148
Mise au point
Le fer est un nutriment contribuant au transport
de l’oxygène et au transfert d’électrons essentiels
à de nombreuses fonctions physiologiques. De
par son implication dans la synthèse de l’hémoglobine
et de la myoglobine, il est indispensable à la mise en
œuvre des échanges gazeux et de la contraction mus-
culaire. Cet ion participe à la défense antibactérienne
des organismes. Il intervient également en tant que
cofacteur dans la régulation de nombreuses enzymes
du métabolisme oxydatif (cytochrome, catalase, myé-
loperoxydase, xanthine oxydase) et de la prolifération
cellulaire (ribonucléotide réductase). En revanche, une
surcharge en fer a des eets délétères liés à l’augmen-
tation du stress oxydatif. Il se produit une inammation
des tissus entraînant une mort cellulaire à l’origine de
dysfonctionnements d’organes ou du développement
de cancers. Le contrôle de l’homéostasie du fer est donc
primordial.
Le cycle du fer
La quantité totale de fer est maintenue entre 2 et 5 g
chez l’adulte. Les besoins quotidiens en fer sont princi-
palement fournis par le recyclage du fer contenu dans
les érythrocytes sénescents (20 mg/j). Il n’existe pas
de système puissant d’élimination du fer, et les pertes
quotidiennes sont limitées à environ 1 mg (urine, sueur,
fèces). Des pertes massives de fer sont toutefois obser-
vées chez la femme au cours des menstruations ou de la
gestation via le placenta. Dans des conditions physiolo-
giques normales, seulement 1 à 2 mg de fer est absorbé
chaque jour au niveau du duodénum et du jéjunum
proximal (gure 1). L’ion Fe3+, présent dans la lumière
de l’intestin, est réduit en Fe
2+
par le cytochrome B fer-
riréductase (DCYTB). Le Fe
2+
peut alors entrer dans les
entérocytes grâce au transporteur transmembranaire
de métal divalent (DMT1) localisé au pôle apical des
cellules. Le fer est stocké dans les entérocytes par liaison
à la ferritine ou exporté par la ferroportine 1 (FPN) située
au pôle basal des cellules. Il est à nouveau oxydé en
Fe3+ par la ferroxydase hephaestine (HEPH) et véhiculé
dans le sang grâce à la transferrine (Tf) jusqu’au site
d’utilisation ou de stockage du fer. La transferrine liée
à deux atomes de fer va se xer sur son récepteur de
haute anité (TfR1) exprimé par les cellules consomma-
trices du fer pour leur fournir les ions nécessaires à leur
activité cellulaire, comme la synthèse de l’hémoglobine
dans les érythroblastes. Le TfR1 interagit avec d’autres
protéines, dont le complexe formé par les protéines de
l’hémochromatose (HFE) et la β2-microglobuline, impli-
qué dans la régulation de l’absorption intestinale du
fer. Au niveau du pôle basal des cellules de la crypte de
l’intestin, le complexe HFE/β2-microglobuline favorise
la captation du fer circulant par stimulation de l’endo-
cytose du TfR1 lié à la transferrine. L’augmentation de
la concentration intracellulaire de fer qui en résulte
L’hepcidine, l’hormone du fer
Hepcidin, the iron hormone
Estelle Louiset*
* Laboratoire de différen-
ciation et communication
neuronale et neuroendocrine,
EA4310, Inserm 413-
IFRMP 413, université de
Rouen, Mont-Saint-Aignan.
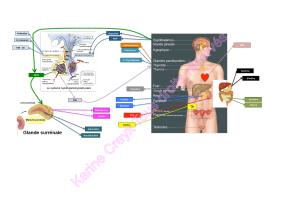
Figure 1. Régulation de l’homéostasie du fer.
L’ion Fe3+ est réduit en Fe2+ par le cytochrome B ferriréductase (DCYTB) avant d’entrer dans les entérocytes via le
transporteur transmembranaire (DMT1). Le fer est stocké dans les entérocytes sous forme de ferritine ou exporté par
la ferroportine 1 (FPN) dans la circulation. Après oxydation par la ferroxydase hephaestine (HEPH), Fe
3+
circule dans le
sang grâce à la transferrine (Tf). Tf-Fe
2
se xe à un récepteur de haute anité (TfR1) exprimé par les érythroblastes et
fournit le fer nécessaire à la synthèse de l’hémoglobine. Le Fe3+ des hématies est recyclé par les macrophages. Tf-Fe2
se lie à la protéine de l’hémochromatose (HFE) des entérocytes pour réduire l’absorption intestinale de fer. Tf-Fe
2
se lie
également à un récepteur de faible anité (TfR2) exprimé par les hépatocytes. Fe3+ est stocké dans les hépatocytes
sous forme de ferritine. Fe3+ contenu dans les hépatocytes ou les macrophages est déversé dans la circulation grâce
à la ferroportine 1 sous le contrôle négatif d’une hormone, l’hepcidine, produite par les hépatocytes. La sécrétion
d’hepcidine est stimulée par Tf-Fe2, HFE, l’hémojuveline (HJV), une infection ou l’IL-6, mais est inhibée par une hypoxie.
➡ : voie principale; ➝ : voie minoritaire.
Hepcidine
Hépatocyte
Fe3+
Fe3+
Fe3+
FPN
FPN
FPN
Fe2+
Fe2+
Fe2+
HEPH
HFE
TfR1 Fe3+
Fe3+
DCYTB
DMT1 Ferritine
Entérocyte
Érythroblaste
Hématies Macrophage
TfR2
Tf
Tf
TfR1
+ Tf-Fe2
+ HFE
+ HJV
+ infection
+ IL-6
– faible O2

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 4 - juillet-août 2009
149
L’hepcidine, l’hormone du fer
entraîne une baisse de l’expression des transporteurs
DMT1 et de la ferroportine 1, ce qui réduit l’absorption
intestinale par les entérocytes. Lorsque le taux circulant
de transferrine saturée est élevé, le complexe Tf-2Fe3+
se lie également à un récepteur de moindre anité, le
TfR2, exprimé par les hépatocytes. Le fer pénètre alors
dans les hépatocytes pour y être stocké sous forme de
ferritine. Le fer contenu dans les hématies est, quant
à lui, recyclé par l’intermédiaire des macrophages, qui
phagocytent les érythrocytes sénescents et dissocient
l’hémoglobine. Le fer est retenu dans les macrophages
circulants ou résidents tels que les cellules de Kuper
du foie. Le fer contenu dans les hépatocytes ou les
macrophages peut être déversé ultérieurement dans
la circulation grâce à la ferroportine 1, en vue d’une
nouvelle utilisation.
Rôle de l’hepcidine
dans l’homéostasie du fer
L’hepcidine est une hormone hépatique de 25 acides
aminés produite par clivage d’un précurseur de haut
poids moléculaire, la préprohepcidine (84 acides ami-
nés) [1]. Le précurseur est codé par le gène HAMP (hep-
cidin antimicrobian peptide), qui compte trois exons
et est localisé chez l’homme sur le chromosome 19.
L’hepcidine est riche en acides aminés basiques et
contient huit cystéines impliquées dans des ponts
disulfures, ce qui favorise la stabilité du peptide dans
le plasma et lui confère une structure en épingle à
cheveux semblable à celle des peptides de la famille
des défensines (gure 2). C.H. Park et al. ont révélé les
propriétés antimicrobiennes de l’hepcidine (2). À ce jour,
il n’existe pas de dosage plasmatique de l’hepcidine.
En revanche, ses métabolites de 20 et 22 acides ami-
nés peuvent être dosés dans les urines, ce qui permet
indirectement d’étudier les mécanismes de contrôle
de la sécrétion d’hepcidine dans des modèles animaux
ou chez l’homme.
C. Pigeon et al. ont montré pour la première fois l’existence
d’un lien entre l’hepcidine et l’homéostasie du fer (3).
En eet, ils ont noté qu’une surcharge en fer stimule
l’expression du gène HAMP chez la souris. Chez l’homme,
il a été observé qu’une élévation du fer sérique s’accom-
pagne d’une augmentation de la concentration des
métabolites de l’hepcidine dans les urines. Par ailleurs,
une injection d’hepcidine provoque chez l’animal une
chute du fer circulant. L’anémie sévère observée chez les
souris transgéniques surexprimant l’hepcidine témoigne
de l’importance physiologique de cette hormone dans
le contrôle de l’homéostasie du fer (4).
L’hepcidine est sécrétée par les hépatocytes en réponse
à une élévation des taux sériques de fer. L’hormone
circulante se lie à la ferroportine 1, le transporteur du fer
qui assure la sortie de l’ion principalement au niveau des
entérocytes et des macrophages, et dans une moindre
mesure au niveau des hépatocytes (gure 1). La liaison
de l’hepcidine à la ferroportine 1 initie l’internalisation
du complexe moléculaire formé, puis sa dégradation
dans les lysosomes, ce qui réduit les flux de fer (5).
L’hepcidine, sécrétée à la suite d’une augmentation des
taux sériques de l’ion, inhibe la sortie du fer des cellules
dans lesquelles il est stocké, ce qui tend à abaisser la
concentration de fer dans la circulation systémique et à
augmenter sa rétention dans les entérocytes, les macro-
Figure 2. Structure de l’hepcidine.
L’hepcidine est riche en acides aminés basiques et compte huit cystéines (A) impliquées dans des ponts disulfures (orange)
qui lui confèrent une structure en épingle à cheveux (B) semblable à celle des peptides de la famille des défensines.
C : des mutations du gène HAMP aectent la région 5’ non codante ou les exons 1, 2 ou 3. D : des mutations ponctuelles
de l’exon 3 modient la structure primaire de l’hepcidine et induisent la perte d’un pont disulfure.
ATG
C70R G71D C78T
R56X
R59G
C70R
G71D
C78T
93delGdel148-151
5’UTR+14 G>A
Exon 1 Exon 2 Exon 3
5’ 3’
A
B
C
D

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 4 - juillet-août 2009
150
Mise au point
phages et les hépatocytes. Il a été rapporté que l’accu-
mulation de fer dans les entérocytes, consécutive à une
injection d’hepcidine, entraîne une baisse de l’expres-
sion des gènes codant DMT1 et DCytB, induisant une
réduction de l’absorption intestinale de fer (6). Il a par
ailleurs été montré qu’une baisse du taux sérique de fer
ou une hypoxie réprime l’expression du gène HAMP (7).
La réduction du taux circulant de l’hormone qui en
découle permet le rétablissement de la ferroportine 1
à la membrane des entérocytes, des macrophages et
des hépatocytes et, par conséquent, la sortie de fer des
cellules de stockage vers la circulation.
Par analogie avec l’insuline, qui maintient l’homéostasie
du glucose en favorisant son stockage sous forme de
glycogène, A. Pietrangelo considère que l’hepcidine,
en induisant le stockage du fer dans le foie sous forme
de ferritine, est l’hormone clé de la régulation de l’ho-
méostasie du fer (8).
Mécanismes de contrôle
de l’expression de l’hepcidine
De nombreuses données indiquent que les protéines
HFE et TFR2, qui jouent le rôle de senseurs du fer en
détectant l’élévation de la saturation de la transferrine,
inuent sur l’expression du gène HAMP par des méca-
nismes non encore élucidés (9, 10). Bien que l’on ne
connaisse pas exactement la fonction de l’hémojuveline
(HJV), on sait que cette protéine membranaire, expri-
mée au niveau du cœur, du muscle squelettique et du
foie, peut être en partie coupée en présence de faibles
concentrations circulantes de fer. Le fragment soluble
résultant du clivage de HJV passe dans la circulation
systémique et est véhiculé jusqu’au foie, où il active
l’expression de l’hepcidine. Ce fragment de HJV est le
facteur stimulateur le plus puissant de la transcription
du gène HAMP (11).
L’analyse de la région promotrice du gène HAMP a révélé
qu’il n’existe pas de séquence consensuelle correspon-
dant à l’élément de réponse au fer (iron response element,
IRE). Le fer n’exerce donc pas un contrôle de la transcrip-
tion du gène HAMP via une iron response protein comme
il le fait pour les gènes codant le DMT1, le récepteur de
la transferrine et la ferroportine 1. En revanche, des sites
de liaison d’autres facteurs de transcription, tels que les
sites CCAAT/enhancer binding protein α (C/EBPα), Smad-
4, HNF4α (hepatic nuclear factor) et USF-2 (upstream
stimulatory factor 2), ont été identiés dans la région pro-
motrice du gène HAMP chez l’homme et les rongeurs.
La combinaison des approches in vitro et in vivo chez
des souris transgéniques a permis de démontrer que
C/EBPα et Smad-4 stimulent, alors que HNF4α réprime
la transcription du gène de l’hepcidine (12, 13). Il a en
particulier été montré que l’accumulation de fer dans
les hépatocytes augmente la production de C/EBPα, qui
stimule à son tour la synthèse d’hepcidine, engendrant
une baisse de l’eux de fer des macrophages et des
entérocytes, ce qui tend à limiter une surcharge hépa-
tique en fer. Bien que le promoteur du gène humain
contienne plusieurs sites consensuels de liaison du
facteur inductible par l’hypoxie (hypoxy induced fac-
tor, HIF), son absence chez les autres mammifères ne
plaide pas en faveur de son rôle dans le contrôle de la
synthèse de l’hepcidine. Les mécanismes impliqués
dans la régulation de l’expression de l’hepcidine ne
sont donc pas totalement élucidés.
Rôle de l’hepcidine dans la pathogénie
de l’anémie due à une inflammation
L’infection et l’inammation chronique sont connues
pour induire une anémie ferriprive. E. Kemna et al. ont
montré qu’une infection bactérienne ou une injection
de lipopolysaccharide engendre une augmentation de
l’expression du gène HAMP (14). En revanche, aucune
modication de l’expression d’hepcidine n’est observée
chez les souris décientes en récepteurs de l’immunité
innée, les récepteurs Toll like de type 4 (TLR4–/–). Ces
travaux révèlent que l’activation des TLR4 présents à la
surface des hépatocytes est à l’origine de la variation de
l’expression d’hepcidine en réponse à une infection. La
réduction du fer sérique qui en découle prive les agents
pathogènes du fer indispensable à leur prolifération.
Cet eet antimicrobien de l’hormone est renforcé par la
capacité de l’hepcidine à perforer la paroi bactérienne
grâce à sa structure en épingle à cheveux, commune
aux défensines (1).
L’inammation aiguë ou chronique qui s’accompagne
d’une production de cytokines pro-inammatoires,
telles que le TNFα, l’IL-1 et l’IL-6, induit également
une augmentation de la synthèse de l’ARNm codant
l’hepcidine. La surexpression de l’hepcidine dans des
modèles animaux de l’inammation est observée chez
les souris décientes en IL-1 et TNFα, mais elle n’est
pas détectée chez les souris dépourvues d’IL-6 (15).
Des expériences réalisées in vitro et in vivo ont conrmé
l’eet stimulateur de l’IL-6 sur la production d’hepcidine
par les hépatocytes. Par ailleurs, une étude clinique a
montré que le blocage de l’action de l’IL-6 par injection
de tocilizumab, un anticorps capable de piéger l’IL-6
circulant, réduit la sécrétion d’hepcidine et améliore
le bilan hématologique des patients sourant d’une

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 4 - juillet-août 2009
151
L’hepcidine, l’hormone du fer
inflammation chronique (16). Les études mécanis-
tiques ont révélé que l’eet stimulateur de l’IL-6 sur la
régulation du gène HAMP est relayé par les facteurs de
transcription smad4 et stat3. Globalement, ces don-
nées indiquent que l’augmentation de la synthèse
d’hepcidine, induite par l’activation des TLR4 et/ou la
production d’IL-6, joue un rôle crucial dans l’établisse-
ment de l’anémie ferriprive au cours de l’infection et/
ou de l’inammation par le biais des actions inhibitrices
de l’hormone sur l’absorption intestinale de fer et la
libération du fer stocké dans les macrophages et les
hépatocytes. La rétention du fer dans les cellules de
stockage induite par l’hepcidine explique également
l’hyperferritinémie observée au cours du processus
inammatoire. E. Nemeth et T. Ganz suggèrent que le
développement d’antagonistes de la ferroportine 1,
le récepteur de l’hepcidine, pourrait fournir des outils
pharmacologiques pour le traitement de l’anémie
observée dans l’inammation chronique (17).
Rôle de l’hepcidine dans la pathogénie
de l’hémochromatose héréditaire
Les mutations inactivatrices de gènes codant des pro-
téines impliquées dans le contrôle de l’homéostasie du
fer engendrent une hémochromatose héréditaire qui
induit une accumulation de fer dans le foie, les arti-
culations, le cœur et les organes endocrines (hypophyse,
pancréas, thyroïde) [tableau I]. Ces mutations aectent
les gènes codant les protéines senseurs du fer, HFE,
TfR2 et HJV, ainsi que l’hepcidine et la ferroportine 1
(gure 3). Sur le plan biologique, la saturation de la
transferrine devient supérieure à 45 % et la ferritinémie
s’élève à plus de 300 µg/l chez l’homme ou 200 µg/l
chez la femme. L’expression clinique de l’hémochro-
matose dépend des atteintes génétiques (tableau II).
Les mutations aectant HFE et TfR2 provoquent des
hémochromatoses modérées d’apparition tardive qui
génèrent principalement un dépôt de fer au niveau
hépatique, alors que celles touchant HJV et l’hepci-
dine sont responsables des formes juvéniles et sévères
de la maladie associées à un hypogonadisme et à des
troubles cardiaques.
L’analyse génétique des mutations du gène HAMP a
révélé que la mutation 54UTR+14G>A de la région 5’
non codante crée, en amont de l’exon 1, un nouveau
codon d’initiation de la traduction qui allonge la taille
du précurseur et induit un décalage de la phase de
lecture. Les délétions observées dans les exons 1 et 2
génèrent aussi des décalages du cadre de lecture. Les
diérentes substitutions ponctuelles de l’exon 3 aec-
Figure 3. Perturbation de l’homéostasie du fer dans l’hémochromatose héréditaire.
Des mutations inactivatrices des gènes codant HFE, l’hémojuveline (HJV), le récepteur de la transferrine (TfR2) ou
l’hepcidine altèrent le fonctionnement d’un des éléments de contrôle de l’homéostasie du fer. Chaque mutation induit
une réduction de la sécrétion d’hepcidine maintenant l’absorption intestinale et le relargage du fer par les macrophages
à des niveaux élevés, ce qui favorise le stockage du fer dans le foie. Des mutations inactivatrices du gène codant la
ferroportine 1 engendrent une perte de sensibilité du récepteur de l’hepcidine empêchant la transduction du signal
inhibiteur de l’hormone sur l’absorption intestinale de fer et sur la sortie des macrophages, ce qui augmente le stockage
hépatique du fer malgré des taux plasmatiques d’hepcidine élevés.
➡ : voie principale; ➝ : voie minoritaire.
Fe3+
Hépatocyte
FPN
Fe2+
TfR1
DCYTB
DMT1
Entérocyte
Hepcidine
Fe3+
Fe3+
Fe3+
FPN
FPN
Fe2+
Fe2+
HFE Fe3+
Ferritine
Érythroblaste
Hématies Macrophage
TfR2
Tf
Tf
TfR1
+ Tf Fe2
+ HFE
+ HJV
– faible O2
Tableau I. Manifestations cliniques des hémochromatoses héréditaires.
Organes Manifestations cliniques
Foie Altération enzymatique, fibrose, cirrhose, hépatocarcinome
Cœur Cardiomyopathie, arythmie
Pancréas Hyperglycémie, diabète
Hypophyse Hypogonadisme, impuissance, aménorrhée
Thyroïde Hypothyroïdisme
Articulations Arthralgie
Peau Pigmentation
Tableau II. Génétique des hémochromatoses héréditaires.
Gène Chromosome Protéine Transmission Âge
d’apparition Manifestations
cliniques
HFE 6p21.3 HFE Récessive 30-50 ans Hépatique
Articulaire
HJV 1p21 Hémojuveline Récessive 20-30 ans Cardiaque
Endocrine
HAMP 19q13.1 Hepcidine Récessive 20-30 ans Cardiaque
Endocrine
TfR2 7q22 Récepteur de la
transferrine 2 Récessive 30-50 ans Hépatique
SLC40A1 2q32 Ferroportine Dominante 40-50 ans Hépatique
Articulaire
HEPH

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XIII - n° 4 - juillet-août 2009
152
Mise au point
Prochain dossier thématique à paraître en octobre 2009
“Comment gérer un diabète lors de troubles cognitifs ?”
Coordination : Anne Vambergue (Lille)
tent la structure primaire de l’hepcidine, supprimant
un pont disulfure et/ou déstabilisant sa structure en
épingle à cheveux (gure 1C). Toutes les mutations du
gène HAMP ont pour conséquence une perte d’expres-
sion de la forme biologiquement active de l’hepcidine.
Les patients porteurs d’une mutation aectant HFE
présentent des taux bas ou normaux d’ARNm du gène
HAMP dans les biopsies de foie et des métabolites de
l’hepcidine dans les urines (18, 19). Les métabolites
urinaires de l’hepcidine sont extrêmement faibles, voire
indétectables, dans les formes juvéniles d’hémochroma-
toses héréditaires (20). De même, un décit en hepcidine
a été observé chez les souris décientes en HFE, TfR2
ou HJV (18, 21). Dans tous ces cas, la faible production
d’hepcidine, qui est anormalement basse au regard de
la surcharge en fer, contribue à maintenir élevés à la fois
l’absorption intestinale de l’ion (8 à 10 mg/j) et son relar-
gage par les macrophages, ce qui favorise le stockage
du fer dans le foie. À l’inverse, il a été rapporté que des
patients porteurs d’une mutation (162delVal) induisant
une perte de fonction du récepteur de l’hepcidine, la
ferroportine 1, présentaient des taux élevés de métabo-
lites urinaires de l’hepcidine (22). Toutefois, l’insensibilité
des récepteurs à l’hepcidine ne leur permettait pas de
maintenir le fer à un niveau physiologique, ce qui a
entraîné l’apparition d’une hémochromatose.
Les patients sourant d’une hémochromatose sont
traités à vie par des saignées d’environ 400 ml répétées
toutes les deux semaines en début de traitement. Ces
saignées, qui visent à réduire le stockage du fer dans
le foie, doivent permettre de faire diminuer le taux de
saturation de la transferrine (< 30 %) et la ferritinémie
(< 50 µg/l). Malheureusement, ces saignées engendrent
une faiblesse chez certains patients, les contraignant à
espacer les ponctions. De plus, les phlébotomies répé-
tées induisent une hypoxie qui renforce l’inhibition de
la production d’hepcidine, aggravant d’autant la sur-
charge en fer. Au vu du décit en hepcidine constaté au
cours des hémochromatoses héréditaires, les patients
qui ne présentent pas une insensibilité à l’hepcidine
pourraient voir leur état fortement amélioré par des
injections d’hepcidine ou par l’absorption de l’un de
ses analogues (8).
Hepcidine et obésité
L’obésité sévère, qui engendre un état inammatoire
caractérisé par une élévation de la CRP, du TNFα et
de l’IL-6 circulants, s’accompagne d’une baisse de la
saturation de la transferrine (68 % des obèses ont un
taux de saturation < 25 %) et d’une anémie (hémo-
globine < 8 mmol/l chez un quart des obèses). Une
étude clinique menée par S. Bekri a montré que le tissu
adipeux sous-cutané des patients obèses exprime anor-
malement le gène HAMP (23). La transcription du gène
codant l’hepcidine dans le tissu adipeux est corrélée à
l’indice de masse corporelle, alors que son expression
hépatique n’est pas aectée par la surcharge pondé-
rale. Les auteurs démontrent également que l’expres-
sion anormale de l’hepcidine dans les adipocytes est
initiée par la production locale d’IL-6. Selon S. Bekri,
l’hepcidine peut être considérée comme une adipokine
dont l’augmentation de la production en cas d’obésité
sévère réduit l’absorption intestinale de fer, engendre
une baisse du fer sérique et une anémie. Les auteurs
recommandent d’inclure un bilan du fer dans le suivi
des patients obèses.
Conclusion
L’hepcidine est une hormone hépatique qui joue un
rôle crucial dans le contrôle de l’homéostasie du fer. Les
mécanismes cellulaires impliqués dans le contrôle de la
sécrétion d’hepcidine ne sont pas totalement élucidés.
Toutefois, on sait que l’hypoxie inhibe sa libération
par les hépatocytes, alors que de nombreux facteurs
comme TLR-4, IL-6, HFE, HJV ou TfR2 la stimulent. La
surexpression ou le décit en hepcidine sont asso-
ciés à des troubles du métabolisme du fer observés
au cours d’une infection, d’une inammation chro-
nique, des hémochromatoses héréditaires ou d’une
obésité sévère. La compréhension des mécanismes de
contrôle de la sécrétion d’hepcidine ouvre de nouvelles
perspectives de traitement des pathologies liées à
des surcharges en fer ou d’une anémie associée à une
inammation.
■
 6
6
1
/
6
100%