REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE

1
REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE
MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE
SCIENTIFIQUE
L’UNIVERSITE
BELKAID- TLEMCEN
FACULTE DES SCIENCES
Laboratoire des Technologies de Séparation et de Purification (LTSP)
DEPARTEMENT DE CHIMIE
Option : Science séparative et environnement
Thème
Présenté par
Mme BENYAHIA Kamila
Soutenu le 20/06/2012
Devant les membres de jury:
Président
M. Didi Mohamed Amine Professeur à
Examinateurs
M. Belkhouche Nasr-Eddine Maitre de conférences
M. Oukabdene Khalil Maitre de conférences
Rapporteur
M. Abderrahim Omar de Tlemcen

2
REMERCIEMENTS
force et les capacités pour réaliser ce travail, et à exprimer ma profonde et sincère gratitude à
tous ceux qui, de près ou de loin
Ce travail a été effectué au Laboratoire des Technologies de Séparation et de Purification
R le professeur
Mohammed Amine Didi, professeur à Ba
travail.
Ma gr
akr Belkaid Tlemcen, je tiens à vous remercier Monsieur d'avoir pris en
charge, avec efficacité et ténacité, le suivi de ce travail, je vous exprime ma profonde
disponibili
de vos relations humaines.
Que le Dr Belkhouche Nasreddine, Dr Oukabdene Khalil reçoivent par ces mots, tous mes
vous prie de croire à ma très
trouvent ici mon profond respect.
témoignée Je vous dois une grande considération
pour votre franche collaboration et votre esprit de convivialité présent dans nos relations le
long de ces études.
oublié.
reconnaissante.

3
SOMMAIRE
Introduction générale
01
Chapitre I
Partie Théorique
A- Technique d’extraction
1. Introduction
03
2. Procédés de séparation
03
3. Les différentes techniques de séparation
03
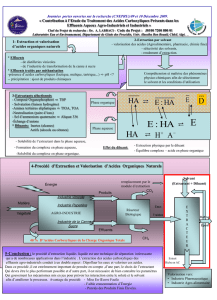
4. Extraction liquide-liquide
04
4.1. Généralités
04
04
4.1.1. a. Classification des extractants
05
06
4.1.2. Le solvant
06
4.1.3. Phase aqueuse
07
-liquide
07
4.3. Principe physico--liquide
08
- liquide
08
4.4.1. Coefficient de distribution
09
10
4.4.3. Aspect cinétique
10
- liquide
11
4.5.1. Extraction par solvatation
11
4.5.2. Extraction par échange cationique
11
4.5.3. Extraction par échange anionique
12
4.5.4. Extraction par solvatation
12
4.5.5. Phénomène de synergisme
13
5. Etude de la complexation des cations métalliques
13
14
14
B- Extractants organophosphorés
1. Introduction
15
2. Aperçu sur le phosphore
15
3. Aperçu général sur les extractants organophosphorés
15
15
5. Les grandes familles des composés organophosphores
16
5.1. Les acides alkyl phosphoniques
16
5.2. Les phosphines
16
5.3. Les esters phosphoriques
16
5.4. Les pyroesters
16
5.5. Les phosphonates
16
C- Métaux lourds
1. Généralités
17
2. Origine de la contamination des sols par les métaux lourds
18
3. Distribution et impact sur
18
3.1. Contamination des sols
18
3.2 Contamination de l'air
19
3.3 Contamination de l'eau
19

4
Le Cadmium
1. Généralités
20
2. Utilisations
20
3. Effet sur la santé
21
ranium
1. Généralités
21
2. Obtention
22
3. Toxicité chimique
22
4. Radiotoxicité
22
uropium
1. Généralités
23
2. Abondance
23
3. Toxicité
23
4. Application
23
Le Thorium
1. Généralités
24
2. Applications
25
3. Toxicité
25
D- Technique d’analyse
1. Introduction
26
26
3. Dosage par spectrophotométrie UV/Visible
26
26
3.1.1. La loi de Beer Lambert
27
3.1.2. Le matériel
29
3.1.2. a. Spectrophotomètres mono-faisceau
29
3.1.2. b. Spectrophotomètres à double faisceau
30
3.2. Dosage du Cd(II) par le PAR
30
3.2.1. Le complexant 4-(2-pyridylazo) resorcinol
30
32
3.2.2. a. Propriétés de l'ArzénazoIII
33
3.2.2. b. Réactions de
33
3.2.2. b.1.Effet du pH sur la complexation
33
Chapitre II
Partie expérimentale
1. Introduction
36
Partie A
36
Partie B
36
Partie C
37
2. Réactifs et produits utilisés
37
3. Instruments et appareils de mesure
37
38
4.1. Analyse du cadmium
38
38
4.3. Analyse de Thorium(IV
38
5. Préparation des solutions
39
5.1. Préparation de la solution mère de CdSO4, (1,0.10-2) mol.L-1
39
5.2. Préparation de la solution mère de UO2(NO3)2 10-2 mol.L-1
39

5
18H39PO3) 10-3mol.L-1
39
5.4. Préparation de la solution de PAR 10-3 mol.L-1
39
10-3 mol.L-1
40
5.6. Préparation de la solution de Th(IV) 10-2 mol.L-1
40
5.7. Préparation de la solution Eu(III) 6,5.10-3 mol.L-1
40
5.8. Préparation de la solution tampon de pH =2,07
40
5.9. Préparation de la solution HCl 9N
40
6. Procédé
40
7. Etude paramétrique
42
7.1. Effet de la concentration initiale
42
42
7.3. Effet de pH
42
7.4. Effet de rapport molaire extractant
42
7.5. Effet de la force ionique
43
7.6. Effet de la température
43
7.7. Comparaison avec les cations Th(IV) et Eu(III)
43
CHAPITRE III
Résultats et discussion
1. Introduction
44
2. Dosage par spectrophotométrie UV/Visible de Cd(II)
44
46
4. Dosage par V)
48
48
6. Extraction du Cadmium(II)
48
6.1. Introduction
48
6.2. Effet du pH de la phase aqueuse
49
52
6.4. Effet de la concentration de Cd(II)
53
53
6.6. Effet du rapport molaire extractant/cadmium
55
6.7. Conclusion sur la partie A
55
56
7.1. Introduction
56
7.2. Effet du pH de la phase aqueuse
56
58
59
60
7.6. Effet du rapport molaire extractant/uranyle
61
7.7. Effet de la force ionique
62
7.8. Effet de la température
63
7.9. Paramètres thermodynamique
64
IV) et de
66
Conclusion générale
68
Bibliographie
70
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
1
/
79
100%