L Perspectives : le traitement des cancers bronchiques de 2011 à 2020

400 | La Lettre du Cancérologue • Vol. XX - n° 6 - juin 2011
DOSSIER THÉMATIQUE
Cancers du poumon
Perspectives : le traitement
des cancers bronchiques
de 2011 à 2020
Perspectives in lung cancer treatment: 2011-2020
D. Planchard*
* Département d’oncologie médicale,
Institut Gustave-Roussy, Villejuif.
L
e traitement des cancers bronchiques non à
petites cellules (CBNPC) a considérablement
progressé ces 25 dernières années, et la chimio-
thérapie et les thérapies moléculaires ciblées ont
désormais un bénéfice limité mais réel. Le début du
e
siècle a été marqué par le développement des
thérapies moléculaires ciblées, qui ont modifié la
prise en charge des patients. L’apport des nouvelles
molécules aux traitements classiques et les progrès
dans la connaissance de la biologie des CBNPC
nous conduisent à une amélioration croissante des
résultats thérapeutiques. Actuellement, la meilleure
démonstration en est la corrélation entre le statut
mutationnel du récepteur du facteur de croissance
épithélial (Epidermal Growth Factor Receptor [EGFR])
et la réponse aux inhibiteurs de tyrosine kinase (ITK)
de l’EGFR (ITK de l’EGFR) et, plus récemment, la corré-
lation du réarrangement ALK (Anaplastic Lymphoma
Kinase) et de la réponse au crizotinib (1, 2). Cela nous
montre que l’identification d’une signature molécu-
laire de la tumeur peut guider notre choix thérapeu-
tique actuel – et plus encore dans les années à venir.
Les données biologiques vont ainsi orienter de plus en
plus nos choix thérapeutiques en identifiant les cibles
les plus pertinentes pour chaque tumeur. L’objectif
des 10 prochaines années est donc la thérapie à la
carte pour chaque patient porteur d’un CBNPC.
Le cancer bronchique :
d’une pathologie d’organe
vers une pathologie biologique
Pendant de nombreuses années, la cancérogenèse
était caractérisée comme le développement d’une
tumeur au sein d’un organe normal. Par la suite, les
cancers bronchiques à petites cellules (CBPC) ont été
séparés des CBNPC, avec divers types histologiques :
les carcinomes épidermoïdes, les adénocarcinomes
et les carcinomes à grandes cellules. Ces dernières
années, de nombreux sous-types tumoraux ont été
individualisés, notamment au sein des adénocarci-
nomes (bronchiolo-alvéolaire, acinaire, papillaire,
etc.). Ces différents sous-types correspondent à des
signatures moléculaires différentes, témoins d’activa-
tion de voies de signalisation intracellulaires propres
à chaque type (EGFR, HER2, FGFR, KRAS, BRAF, PI3K/
AKT, etc.). Cette individualisation moléculaire est
récente et fait suite à celle de populations bénéficiant
des ITK de l’EGFR, la première étant celle des sujets
non fumeurs présentant un profil oncogénique parti-
culier (3). La vision du cancer apparaît donc comme
celle d’une maladie du génome (4, 5). Des aberra-
tions génomiques complexes visant de multiples
gènes, secondaires à des mutations, des anomalies
du nombre de copies ou des modifications épigé-
nétiques aboutissent dans la plupart des tumeurs
à des modifications des voies de signalisation qui
déterminent le comportement de la cellule cancé-
reuse et du devenir des patients. Les technologies
modernes de génomique et d’épigénomique modifient
considérablement notre compréhension des cancers,
leur caractérisation et l’approche des traitements
adaptés à la biologie. L’analyse génétique permet
ainsi l’identification de facteurs prédictifs du bénéfice
thérapeutique pour l’obtention d’une réponse théra-
peutique optimale et d’une moindre toxicité. Cette
approche à la carte nous entraîne de plus en plus
vers la possibilité d’individualiser le traitement selon
le profil moléculaire des patients (4). Plusieurs tests
moléculaires génomiques validés, réalisés à partir du

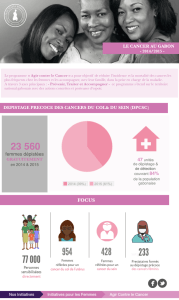
Inconnu (33 %)
Mutations de KRAS b
Mutations de l’EGFR a
Mutation de LKB1
Mutation de PI3K c
a Sensible aux inhibiteurs EGFR.
b Résistant aux inhibiteurs EGFR.
c Sensible aux inhibiteurs de PI3K.
d Sensible aux inhibiteurs d’HER2.
e Sensible aux inhibiteurs d’ALK.
ALK : Anaplasic Lymphoma Kinase ;
EGFR : Epidermal Growth Factor Receptor ;
HER2 : Human Epidermal growth factor Receptor-2 ;
PI3K : phosphoinositide 3-kinase.
Mutation de HER2 d
Amplification de Met b
Fusion de ALK e
Figure 1. Anomalies moléculaires clés
dans le CBNPC, pouvant influencer la
prescription de thérapies ciblées (4).
La Lettre du Cancérologue • Vol. XX - n° 6 - juin 2011 | 401
Résumé
Il existe dans le CBNPC une meilleure compréhension de la biologie moléculaire permettant un ciblage
thérapeutique intelligent des patients en fonction des altérations moléculaires.
Les principales altérations moléculaires connues à ce jour dans le CBNPC sont les mutations d’EGFR et de
KRAS, les translocations EML4-ALK, les amplifications de MET, HER2 et FGFR1 auxquelles correspondent
des thérapies ciblées potentielles.
De nombreuses anomalies moléculaires supplémentaires sont régulièrement mises au jour avec de nombreuses
nouvelles molécules en cours d’essais thérapeutiques.
Le tissu tumoral reste à ce jour la clé de l’analyse moléculaire et du ciblage thérapeutique avec le déve-
loppement d’analyses sur de la cytologie et des cellules tumorales circulantes.
Mots-clés
Cancer bronchique
non à petites cellules
Thérapies ciblées
EGFR
ALK
ERCC1
Summary
There is a better understanding
of biology disease in NSCLC
allowing personalized therapy.
This offer the potential of iden-
tifying patients with those
tumour types considered most
likely to benefit from “molecu-
larly targeted therapies”.
The most frequent molecular
alterations in NSCLC are the
EGFR, and KRAS mutations,
the EML4-ALK translocation,
the MET, HER2, and FGFR1
amplifications with potential
targeted therapies. Numerous
additional abnormalities are
regularly updated with new
drugs tested in clinical trials.
The tumoral tissue remains
the key of molecular analysis.
Keywords
Non-small-cell lung cancer
Target therapy
EGFR
ALK
ERCC1
tissu tumoral, font maintenant partie de la prise de
décision thérapeutique pour de nombreux cancers,
dont celui du poumon (2, 6). Cependant, le champ
des nouvelles thérapies à la carte comporte beaucoup
de défis, avec un niveau d’échec thérapeutique qui
reste important, des difficultés d’identification des
cibles, de validation de biomarqueurs, et des méca-
nismes de résistances inconnus. L’achèvement du
séquençage du génome humain, associé aux déve-
loppements technologiques majeurs, a ouvert la voie
à une analyse complète des déterminants génétiques
des cancers bronchiques. Dans le cadre de la cancéro-
logie, les questions à résoudre sont complexes, car il
faut prendre en compte non seulement les variations
génétiques propres à chaque individu, mais aussi les
altérations acquises du génome tumoral ainsi que
la génomique fonctionnelle ; celle-ci vise en effet
à déterminer la fonction et l’expression des gènes
en caractérisant le transcriptome et le protéome,
donnant accès à des notions de perturbation des
réseaux génétiques, indispensables à la compréhen-
sion des conséquences fonctionnelles des anoma-
lies chromosomiques (5). Les techniques modernes
passent par l’étude de l’expression haut débit du
génome, transcriptome, protéome… qui nécessite
l’utilisation de l’informatique et de la bio-informa-
tique pour accélérer la phase d’analyse complexe de
multiples données. De plus, il va être facile d’obtenir
la séquence complète d’un génome humain, et ce à
des coûts abordables. Le défi à venir est donc d’inté-
grer le développement des technologies “omiques”
(génomique, protéomique, métabolique, etc.) haut
débit qui étudient les tumeurs au niveau de l’ADN, de
l’ARN, de la protéine et du métabolite. La découverte
de la coordination de ces événements constituera
un moment crucial dans le processus du dévelop-
pement des thérapies ciblées, qui vise à trouver le
“talon d’Achille” de la dépendance oncogénique de
chacune des tumeurs et ainsi à pouvoir induire une
“mortalité synthétique”.
Évolution vers de nouvelles
cibles et classification du CBNPC
L’identification d’une voie d’addiction oncogénique
au niveau tumoral permet ainsi de reclasser les
CBNPC non plus en fonction d’une classification
histologique mais en fonction d’une classification
moléculaire. Cela a pour implication de proposer
une thérapie adaptée à une anomalie moléculaire,
en fonction du profil mutationnel de la tumeur (diffé-
rentes validations sont en cours). Si l’on considère
les principales altérations moléculaires connues dans
le CBNPC (mutations d’EGFR [10 %], translocation
EML4-ALK [4 %], amplification de Met [3 %], ampli-
fication de HER2 [1 à 2 %], amplification de FGFR
[3 %], mutations de KRAS [20 à 30 %]) auxquelles
correspondent des thérapies ciblées potentielles,
l’on s’aperçoit qu’il est actuellement possible de

402 | La Lettre du Cancérologue • Vol. XX - n° 6 - juin 2011
DOSSIER THÉMATIQUE
Cancers du poumon Perspectives : le traitement des cancers bronchiques de 2011 à 2020
cibler environ 30 % des CBNPC (figure 1, p. 401) [4].
L’objectif des 10 prochaines années est donc claire-
ment la découverte et la validation d’autres anoma-
lies ou cibles thérapeutiques dans l’espoir de parvenir
à laisser l’ensemble des CBNPC. Le CBNPC ne sera
alors plus une pathologie d’organe mais une patho-
logie relevant d’anomalies biologiques et il s’agira
alors d’un ensemble de maladies rares. Aujourd’hui,
avec les progrès de la biologie et de la biochimie, il
est relativement facile de créer des molécules visant
une anomalie moléculaire déterminée. Ce qui, en
revanche, constitue une gageure est l’identification
des cibles moléculaires responsables des addictions
oncogéniques. Le véritable défi réside donc dans
nos capacités actuelles et futures à disposer des
outils biologiques nécessaires à l’identification de
ces cibles.
Étudier le nombre de copies de l’ADN
Les aberrations du nombre de copies de l’ADN indui-
sent une modification de la quantité et, ainsi, une
modification de l’organisation du matériel géno-
mique, ce qui peut conduire à une augmentation ou
à une diminution de l’activité transcriptionnelle de
gènes clés ou de l’ARN régulateur. Ces aberrations
du nombre de copies d’ADN peuvent être à l’origine
de la modification d’un seul gène ou affecter une
région chromosomique plus importante comportant
plusieurs gènes. Ces aberrations du nombre de copies
de l’ADN sont soit héréditaires, soit causées par des
anomalies somatiques telles que les délétions, dupli-
cations, inversions ou translocations. De nombreux
gains (1q31, 3q25-27, 5p13-14, 8q23-24, etc.) ou
pertes de chromosomes ou de fragments de chro-
mosomes (3p21, 8p22, 9p21-22, 13q22, 17p12-13,
etc.) sont retrouvés dans les CBNPC (5). Les techno-
logies à haut débit incluent différentes techniques
– comme l’hybridation génomique comparative, le
caryotype numérique, les microarrays d’oligonucléo-
tides (Representational Oligonucleotide MicroArray
[ROMA]), les microarrays de polymorphismes nucléo-
tidiques simples, les sondes moléculaires d’inversion
ou le séquençage de nouvelle génération – qui sont
maintenant capables de détecter rapidement et effi-
cacement des modifications du nombre de copies à
travers un génome tumoral entier.
Jusqu’à récemment, les réarrangements chromo-
somiques, comme les translocations, ne faisaient pas
partie du tableau des anomalies chromosomiques
des CBNPC. Or, il est clair aujourd’hui qu’une trans-
location comme ALK-EML4 (étude par hybridation
in situ en fluorescence ou fluorescence in situ hybri-
dization [FISH]) est une voie oncogénique impor-
tante dans certains CBNPC (4 %) avec un ciblage
moléculaire possible (2).
Étudier la présence de mutations
de l’ADN
La présence de mutations au niveau de certains gènes
représente actuellement le critère le plus prédictif à
la fois du risque de développer certains cancers et
de réponse à certaines thérapies ciblées (1, 6). Cette
analyse est relativement simple par séquençage du
gène cible permettant de déterminer la présence
ou l’absence de la mutation d’un gène. Cette étude
génomique paraît d’interprétation plus aisée que celle
du transcriptome ou du protéome (7). L’enjeu des
années futures va donc être de déterminer si l’éva-
luation de diverses mutations est le reflet ou non
d’une meilleure compréhension de la biologie cellu-
laire et si elle constitue un meilleur facteur prédictif
pour l’adaptation thérapeutique que l’analyse de
la génomique fonctionnelle (6). Cela est probable-
ment vrai lorsque la mutation isolée concerne un
oncogène majeur de l’oncogenèse dont l’expression
de la protéine “anormale” constitue un “driver” du
processus tumoral. L’exemple par excellence est celui
que fournissent les mutations du gène de l’EGFR, dont
l’inhibition seule par les ITK de l’EGFR suffit à bloquer
efficacement l’oncogenèse tumorale (1). Le deuxième
exemple est celui du gène ALK et de l’inhibition de la
protéine de fusion ALK-EML4 par le crizotinib (2). Cela
est sans doute aussi le cas pour d’autres anomalies
génomiques, le bénéfice de leur inhibition restant par
ailleurs à valider (mutations de KRAS, HER2, PI3K,
etc.). À l’inverse, d’autres mutations vont ne participer
qu’à la prolifération, à la croissance, à l’invasion, à
l’angiogenèse… tumorales, sans être de véritables
“drivers” de l’oncogenèse. Dans ce cas, l’analyse
génomique devra être plus complexe (séquençage à
haut débit) et devra s’intéresser à l’étude de plusieurs
milliers de gènes pour en dégager des profils (8).
Étudier le profil épigénétique
Les modifications épigénétiques liées à des anoma-
lies de la méthylation sont des événements fréquents
dans les CBNPC (9). L’inactivation génique par
méthylation dans des régions promotrices est
fréquente dans les CBNPC (par exemple l’inactiva-
tion par méthylation des gènes CDKN2A, MGMT,

404 | La Lettre du Cancérologue • Vol. XX - n° 6 - juin 2011
DOSSIER THÉMATIQUE
Cancers du poumon Perspectives : le traitement des cancers bronchiques de 2011 à 2020
DAPK, TIMP-3, APC, RASSF1A, etc.). Il est alors
possible de cibler cette méthylation par des théra-
pies. À l’inverse, la déméthylation de régions promo-
trices de gènes normalement inhibés au cours du
développement serait la cause de la réexpression
de gènes embryonnaires, comme MAGE dans les
CBNPC.
Un certain nombre de techniques pour l’évalua-
tion de l’état de méthylation des gènes du génome
(méthylome) sont en cours de développement.
Comme cela est le cas pour d’autres technologies,
les approches de séquençage de dernière généra-
tion devraient supplanter les technologies actuelles.
Restent des défis : la validation des observations et
le développement d’approches biologiques pour
déterminer la pertinence fonctionnelle des modifi-
cations du statut de méthylation de certains gènes.
Étudier le profil d’expression des gènes
La capacité à mesurer des milliers de transcrits
d’ARNm dans une simple analyse a abouti à une
augmentation rapide de notre compréhension de
la pathophysiologie tumorale, qui nous a permis
de nous orienter vers une classification molé-
culaire des tumeurs et l’établissement de profils
transcriptionnels pronostiques et prédictifs (10).
L’évaluation de profils transcriptionnels fournit la
possibilité d’isoler des groupes pronostiques diffé-
rents parmi les patients opérés d’un CBNPC et de
déterminer le risque de récidive et l’intérêt d’un
traitement adjuvant (11). De la même manière, il
est a priori possible de déterminer des profils prédic-
tifs de réponse aux molécules de chimiothérapie
et thérapies ciblées (12, 13). Il est aussi possible de
classer les tumeurs en fonction du profil transcripto-
mique (14). Un certain nombre de tests génomiques
sont en cours d’évaluation aux différents stades de
la maladie, notamment pour évaluer la capacité
de prédiction du bénéfice des thérapies ciblées en
fonction de l’activation des voies de signalisation.
Par ailleurs, l’étude de l’expression des microARN
(fragments d’ARN non codants [miARN]) paraît de
plus en plus importante. Les miARN interviennent
dans la régulation des gènes et leur implication dans
le cancer sont actuellement – et depuis quelques
années – l’objet d’une importante activité de
recherche (15). Il est vraisemblable que des straté-
gies thérapeutiques ciblées (via des oligonucléotides
interagissant avec leurs messagers cibles) sur un
ou quelques miARN puissent voir le jour dans les
prochaines années.
Étudier l’expression protéique
Les techniques traditionnelles d’étude des protéines,
comme le Western-Blot ou l’Enzyme Linked Immu-
noSorbent Assay (ELISA), ne permettent d’étudier
l’expression et la phosphorylation que d’un nombre
limité de protéines et ne donnent pas la possibilité
de dresser une cartographie complète des différentes
voies de transduction du signal intracellulaire. Des
systèmes d’arrays haut débit (Reverse Phase Proteins
Arrays [RPPA], etc.) permettent d’étudier un plus
grand nombre de protéines candidates avec peu de
matériel protéique nécessaire (10). Compte tenu
du fait que la plupart des marqueurs moléculaires
et des cibles thérapeutiques sont des protéines, la
détermination du profil protéique apparaît poten-
tiellement comme l’une des meilleures évalua-
tions de la fonctionnalité et de la pharmacologie
cellulaire, notamment comparativement au profil
transcriptionnel. Les systèmes d’évaluation à haut
débit peuvent mesurer simultanément l’activation,
la prolifération, l’apoptose, ou n’importe quel autre
processus cellulaire pour lequel des anticorps spéci-
fiques existent. La validité clinique et les avantages
potentiels de tels systèmes sont en cours de valida-
tion, et leurs résultats sont prometteurs.
La clé du progrès : le matériel
tumoral (modèles cellulaires,
biopsies tumorales)
Il est important de pouvoir travailler à partir
d’échantillons tumoraux de patients. Cependant,
les modèles utilisant des lignées cellulaires sont
un outil précieux pour nous aider à avancer dans
l’identification de nouvelles cibles thérapeutiques. Le
succès des thérapies antitumorales est dépendant de
la compréhension minutieuse des interactions à un
niveau moléculaire (sensibilité/résistance). En dehors
de la compréhension mécanistique, l’autre approche
sur le plan cellulaire est représentée par le scree-
ning de molécules au travers de lignées cellulaires,
représentatives de différents types d’aberrations
génomiques et protéiques (11). Il est possible d’ima-
giner que, dans les années à venir, nous disposerons
d’un système d’analyse des différentes molécules
sur lequel il suffira de déposer des cellules tumorales
issues de la tumeur primitive du patient afin d’en
déterminer la sensibilité tumorale in vitro.
Au-delà des modèles cellulaires, il nous faut savoir
explorer les données cliniques et biologiques des

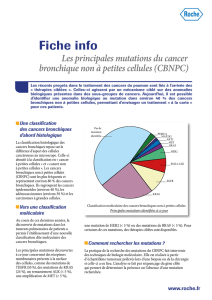
Figure 2. Les différentes voies de réparation de l’ADN.
Erreurs
réplication
ADN
Lésions
des bases
Adduits intrabrins
Adduits volumineux
Systèmes de réparation de l’ADN
Cassures
double-brin
Adduits d’alkyles
bases d’ADN
(mésappariements)
A-G, T-C ERCC1
TFIIH
RPA
XPF
XPA
XPG
XPC
ATM
Rad51
BRCA1
BRCA2
DNA-PK
Ku70, Ku80
MRN
XRCC4
AGT
MSH2-MSH6
MLH1-PMS2
Glycosylase
APE1
PARP
MMR BER NER HR NHEJ DR
BER : Base Excision Repair ; BRCA1 : BReast CAncer 1 ; DR : Direct Repair ; ERCC1 : Excision Repair Cross-Complementation
group 1 ; HR : Homologous Repair ; MMR : Mismatch Repair ou réparation des mésappariements ; NHEJ : Non-Homologous
End-Joining ; NER : Nucleotide Excision Repair ; PARP : Poly (ADP-Ribose) Polymerase.
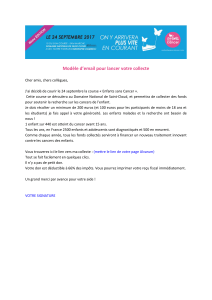
Figure 3. Étude de phase II (TASTE) de stratégie selon le statut EGFR et ERCC1 en adju-
vant chez des patients opérés d’un CBNPC non épidermoïde de stades II et IIIA (non N2).
Bras contrôle
Cisplatine + pémétrexed
EGFR
muté Erlotinib
ERCC1+ Observation
ERCC1– Cisplatine + pémétrexed
R
Bras expérimental
EGFR
non muté
EGFR ; Epidermal Growth Factor Receptor ; ERCC1 : Excision Repair Cross-Complementation group 1 ; R : randomisation.
La Lettre du Cancérologue • Vol. XX - n° 6 - juin 2011 | 405
DOSSIER THÉMATIQUE
patients bénéficiant de nouvelles thérapies dans
le cadre d’essais cliniques. Un certain nombre de
patients (généralement faible mais indéniable) inclus
dans les essais précoces (phases I/II) présentent un
bénéfice clinique avec une réponse tumorale qui
peut être majeure. L’analyse tumorale de ces patients
doit nous aider à comprendre les mécanismes sous-
jacents du bénéfice thérapeutique et à identifier des
biomarqueurs des thérapies. Les nouvelles technolo-
gies nous permettront sans doute de mieux caracté-
riser ces tumeurs et de définir l’aberration moléculaire
qui sous-tend leur sensibilité thérapeutique. Ainsi, des
études sur un petit nombre de patients nous mènent
à la démonstration, par exemple, que la présence
de mutations de l’EGFR et celle d’une translocation
ALK-EML4 identifient des populations de patients
sensibles respectivement aux ITK de l’EGFR ou au
crizotinib. Ces résultats soulignent l’importance et
la nécessité primordiale qu’il y a à disposer de tissu
tumoral récent pour les analyses biologiques.
Ciblage des voies
de la réparation de l’ADN
Les cellules disposent d’au moins six voies différentes
de la réparation de l’ADN : réparation directe (Direct
Repair [DR]), réparation par excision de nucléotides
(Nucleotide Excision Repair [NER]), réparation par
excision de base (Base Excision Repair [BER]), répara-
tion des mésappariements (Mismatch Repair [MMR]),
réparation homologue (Homologous Repair [HR]) et
réparation par recombinaison non homologue (Non-
Homologous End-Joining [NHEJ]) [figure 2]. Chacun
des mécanismes de la réparation est “spécialisé”
dans la prise en charge d’un ou de plusieurs types
de lésion de l’ADN. De manière générale, c’est la
forte capacité de la cellule à réparer les dommages
qui entraîne une baisse de l’instabilité génétique
dans la cellule cancéreuse (facteur pronostique favo-
rable en l’absence de traitement). À l’inverse, une
altération des fonctions de réparation conduit à la
mort cellulaire face aux dommages de l’ADN (facteur
prédictif favorable de réponse au traitement). Les
biomarqueurs moléculaires de la réparation de l’ADN
semblent être l’une des voies de la sélection théra-
peutique ainsi qu’une cible potentielle. L’Excision
Repair Cross-Complementation group 1 (ERCC1) [voie
de réparation du NER] semble actuellement être
l’un des biomarqueurs les plus prometteurs pour
prédire le bénéfice d’une chimiothérapie à base de
sel de platine (16). En effet, l’absence ou un faible
niveau d’expression de la protéine ERCC1 prédit le
bénéfice d’une chimiothérapie adjuvante à base de
sels de platine des CBNPC opérés et, à l’inverse, un
fort niveau d’expression d’ERCC1 en prédit l’absence
de bénéfice. Une confirmation de l’utilité d’ERCC1
dans des études prospectives est nécessaire afin
d’envisager son utilisation en routine clinique.
C’est pourquoi une étude prospective française de
phase II/ III est actuellement en cours pour confirmer
ERCC1 comme biomarqueur prédictif du bénéfice
d’une chimiothérapie adjuvante à base de sel de
platine (étude TASTE [TAilored post-Surgical Therapy
in Early stage NSCLC], sous l’égide de l’Intergroupe
francophone de cancérologie thoracique [IFCT])
[figure 3]. D’autres études au stade métastatique
sont également menées. Le deuxième biomarqueur
actuellement très “médiatisé” (notamment dans
 6
7
8
6
7
8
1
/
8
100%