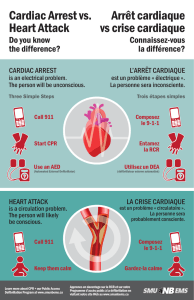
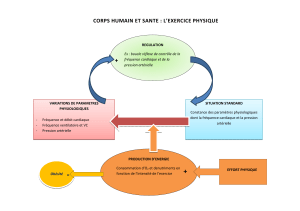
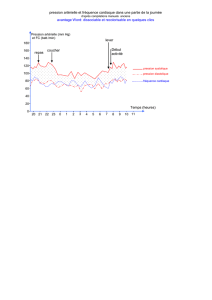
Effets de 6 semaines d`entraînement par intervalles à haute intensité

Effets de 6 semaines d’entraînement par intervalles à
haute intensité sur la fonction cardiaque de repos, la
pression artérielle ambulatoire et la variabilité de la
fréquence cardiaque chez des athlètes masculins
d’endurance
Mémoire
Olivier Le Blanc
Maîtrise en kinésiologie
Maître ès sciences (M. Sc.)
Québec, Canada
© Olivier Le Blanc, 2015


iii
Résumé
Il existe encore plusieurs interrogations par rapport à la fonction cardiaque de repos chez l’athlète
d’endurance. L’entraînement par intervalles à haute intensité (HIIT) est de plus en plus populaire chez les
athlètes, mais ses effets sur la fonction cardiaque de repos demeurent encore inconnus. Le but de cette étude
fut donc d’étudier les effets de six semaines d’HIIT sur la fonction cardiaque de repos, la pression artérielle
ambulatoire (PAA) et la variabilité de la fréquence cardiaque (VFC) chez l’athlète de sport d’endurance. Après
six semaines d’entraînement, les structures et fonctions du ventricule gauche (VG) ainsi que la VFC sont
restées similaires. L’HIIT a diminué la PAA. En conclusion, ces résultats suggèrent que six semaines d’HIIT
n’affectent pas la fonction cardiaque de repos, ni la VFC, mais permettent de diminuer la PAA d’athlètes
d’endurance.


v
Abstract
There is still a lot of interrogations regarding the mechanisms underlying the resting cardiac function
adaptations of endurance athletes. High intensity interval training (HIIT) seems to be the new trend in this
population. The impact of this training method on resting cardiac function in endurance athletes is still
unknown. The purpose of this study was to investigate the impact of six weeks of HIIT on the cardiac function
at rest, ambulatory blood pressure monitoring (ABPM) and heart rate variability (HRV) in endurance athletes.
Following six weeks of HIIT, no sign of ventricular dilatation nor hypertrophy in the left ventricle (LV) were
observed. Diastolic and systolic functions were preserved following training. HIIT decreased ABPM, reflected
by a lowered systolic (SBP), mean (MAP) and pulse (PP) pressures. HRV remained at baseline level. These
results suggest that six weeks of HIIT does not affect cardiac function at rest, nor HRV, but reduces ABP.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
1
/
130
100%