File - L3 Bichat 2013-2014

!"#$%&'&()*+,$#$
-#.//.-0/1$23104/0310$
56$78)9,$:;,<=>9+?4@A'?+B$
C;'&;(B=,+<,$D$%,EA39)'?4F3,6G3HI,$
C;'&;(9,G(6)G,$D$J;+*+,6,9$FB6),99,$$
$
$
$
$
$
$
!"#$%&'&(&&)&*+,+-./%&+#0"%"*.1#/%&
$/!/%%.2/%&
$
$
$
$
$
$
$
$
!
"#$%&'!()*(!+),%&!-&!-)((.&,(/!0$(!01.'.2*&(!
34.'56,75!-&!0&55&!%$5.8,&!&(5!-&!0)%9,&'-,&!1&!,$.()''&%&'5!:!9$(!-&!-65$.1(!;!($<).,!(*,!
1&(!%$1$-.&(!%&'5.)''6&(!=!!
$
$
$

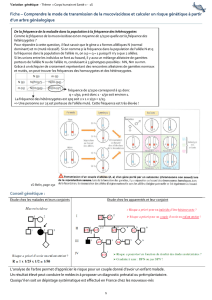
.3 *454678&9:;:<7=>8&?8;6:578;;8&68&<@4;A?7AA7B;&
4><BAB?7=>8&@:C8AA7D8&E*+$F&
&
KL %&'&6A9)(&<$
CA==,9<$D$
! 9,<$3&(&6;MB?;(,<$<;'($<A)'<$$
! <,+9<$9,<$3;E;MB?;(,<$,N=6)E,'($9A$EA9A8),$
FA6AG(&6)<()*+,<$D$OP$6,G3,6G3,6$<+6$+'$$,>,&!?6'6$1)?.2*&Q$$
4 9,<$<+R,(<$A((,)'(<$'A)<<,'($8,$=A6,'(<$';6EA+N$EA)<$3&(&6;MB?;(,<$D$9A$=6;=;6();'$
,'>A'(<$A((,)'(<$,<($8,$S$,($G,99,$8,<$,'>A'(<$3&(&6;MB?;(,<$8;'G$(6A'<E,((,+6<$
,<($8,$TL$$
4 UA$EA9A8),$(;+G3,$8,$>AV;'$&?A9,$9,<$8,+N$<,N,<$
4 U,<$<+R,(<$A((,)'(<$<,$6,(6;+I,'($9,$=9+<$<;+I,'($8A'<$9A$EWE,$>6A(6),$$$D$
6&=A6()();'$3;6)M;'(A9,L$
4 !'$,NGH<$8,$EA6)A?,$G;'<A'?+)'$A+?E,'(,$9A$>6&*+,'G,$8,$G,<$EA9A8),<L$$
$
:L J6&*+,'G,$8,<$XKC$
56,';'<$8,+N$)'8)I)8+<$3&(&6;MB?;(,<$
=;+6$9YA99H9,$KZ$*+)$<,6;'($K/.K-$,($
G;'<)8&6;'<$K-$G;EE,$9YA99H9,$8&9&(H6,L$
U,$=H6,$=;+66A$(6A'<E,((6,$8A'<$<,<$
?AEH(,<$<;)($K/$<;)($K-Z$8,$EWE,$=;+6$
9A$EH6,L$7'$A+6A$8;'G$<;)($$
4 +'$,'>A'($K/.K/$Z$3;E;MB?;(,$
<A)'$[$S$$
4 +'$,'>A'($K-.K-Z$3;E;MB?;(,$
A((,)'($[$S$$
4 +'$,'>A'($K/.K-Z$$3&(&6;MB?;(,$
<A)'$[$T$$
$
F,9A$';+<$=,6E,($&?A9,E,'($8,$0$10*1&,!1$!+,62*&'0&!-&(!@AB$?6\G,$P$9A$>6&*+,'G,$8,<$
A99H9,<$$
4 =$[$>6&*+,'G,$8,$K/$
4 *$[$>6&*+,'G,$8,$K-$D$A99H9,$E+(&$$
4 =]*[/$O)9$'YB$A$*+,$-$>;6E,<$A99&9)*+,<Q$$$$$$$
! G@:=>8;C8&68&54&?454678$^$8A'<$9A$=;=+9A();'$,<($=H$$
! G@:=>8;C8&68A&I:<:@BJK9B<8A$,<($HL=&Z$,($<)$*$__$/$Z$A9;6<$-=*$[$-O/4*QN*$[$-*$
! G@:=>8;C8&68A&IB?BJK9B<8A&A47;A&8A'<$9A$=;=+9A();'$,<($LH&$
! G@:=>8;C8&9:;B<KL7=>8&8,$G,<$A99H9,<$D$=H&M&HL=&M&LH&N&O$$
UY)'(&6W($,<($8,$GA9G+9,6$9A$>6&*+,'G,$8,<$XKCZ$*+)$G;66,<=;'8$P$9A$>6&*+,'G,$8,<$
3;E;MB?;(,<$A((,)'(<Z$A)'<)$*+,$9,$6)<*+,$8,$(6A'<E)<<);'$A99H9,$E+(&$A+$<,)'$8Y+'$G;+=9,Z$
G,$*+)$G;66,<=;'8$$P$9A$>6&*+,'G,$8,<$3&(&6;MB?;(,<L$K)'<)Z$L5>A&54&?454678&8A<&@4@8P&
L5>A&54&G@:=>8;C8&68A&I:<:@BJK9B<8A&8A<&G47Q58R&&
Fréquence des maladies récessives autosomiques
A2
A1
A
A1A1
A2A2
A2A1
A1A2A1
A2
A1
A2
A1A2 A1A2
A1A1 A2A2
A1A2A1A2
¼= 25%
homozygotes
A2
½= 50%
hétérozygotes
A1A2
si A2 = allèle délétère
maladie autosomique récessive :
fréquence de la maladie Q = q2
fréquence hétérozygotes Q = 2pq= 2(1-q)q
si q est petit /1 = 2q
fréquence génotypique
=
q
2
+ 2pq + p
2
p= fréquence allèle A1
q= fréquence allèle A2
p + q = 1
p = 1- q
¼= 25%
homozygotes
A1

FL F;'<A'?+)')(&$,($XKC$$
F,9A$G;66,<=;'8$P$8,<$)'8)I)8+<$*+)Z$ABA'($+'$A'GW(6,$G;EE+'Z$<;'($=9+<$P$6)<*+,$8,$
=6&<,'(,6$+',$3;E;MB?;(),L$5A6$G;'<&*+,'(Z$=9+<$)9$B$A+6A$8YA99H9,<$,'$G;EE+'$,($=9+<$9,$
6)<*+,$8,$XKC$A+?E,'(,$G3,M$9Y,'>A'(L$F,$6)<*+,$IA$8)6,G(,E,'($8&=,'86,$8,$';E`6,<$
8,$G3A)';'<$<&=A6A'($9Y,'>A'($A((,)'($,($<,<$-$=A6,'(<$3&(&6;MB?;(,<$$8,$9YA'GW(6,$
G;EE+'$O-&?,6!-&!9$,&'56Q$D$9;?)*+,E,'(Z$=9+<$9YA'GW(6,$G;EE+'$,<($=6;G3,Z$=9+<$9,$
G;,>>)G),'($8,$G;'<A'?+)')(&$,<($&9,I&$,($$8;'G$=9+<$9,$6)<*+,$8Y3;E;MB?;(),$8,$9YA99H9,$
E+(&$,<($&9,I&L$$
K)'<)Z$9A$G;'<A'?+)')(&$$
! 4>9?8;<8&58&@7A=>8&68&*+$$GP8$9A$>6&*+,'G,$8,$9Y3;E;MB?;(),$=A6$A<G,'8A'G,L$$
! 'YA+?E,'(,$L4A$9,$6)<*+,$8,$EA9A8),<$8;E)'A'(,<L$
! $',$E;8)>),$L4A&@7A=>8&68&?><4<7B;A&;7&58>@&G@:=>8;C8R&
! =9+<$9A$EA9A8),$,<($6A6,Z$,($=9+<$9A$G;'<A'?+)')(&$<,$>A)($6,<<,'()6$$
$
J6&*+,'G,$8,$9A$EA9A8),$D$$
a)$*$,<($9A$>6&*+,'G,$8,$9A$E+(A();'$8A'<$9A$=;=+9A();'$?&'&6A9,$,($*-$9A$>6&*+,'G,$8,$9A$
EA9A8),$D$9A$>6&*+,'G,$8,$9A$EA9A8),$,'$GA<$8,$G;'<A'?+)')(&$,<($G=&EGN&G4C<8>@&68&
CB;A4;9>7;7<:FR&$5A6$,N,E=9,Z$>[/./b$,'(6,$G;+<)'<$?,6EA)'<L$$
$
cL$=A6()G+9A6)(&<$E;9&G+9A)6,<$8,<$XKC$$
$
/L d&(&6;?&'&)(&$?&'&()*+,$$
UY3&(&6;?&'&)(&$?&'&()*+,$<,$6,'G;'(6,$9;6<*+,$9A$?S?8&?454678$O=3&';(B=,Q$=,+($
W(6,$8+,$P$L5>A78>@A&9T;8AR$
"e$D$C&()';=A(3),<$=)?E,'(A)6,<$D$=9+<$8,$f0$?H',<$,'$GA+<,$4g$8)>>)G+9(&<$6,G3,6G3,$8,<$
A';EA9),<$E;9&G+9A)6,<L$$
-L d&(&6;?&'&)(&$A99&9)*+,$$
UY3&(&6;?&'&)(&$A99&9)*+,$<,$6,'G;'(6,$9;6<*+,$8,$;B?Q@8>A8A&?><4<7B;A$8Y+'$?S?8&
9T;8$<;'($6,<=;'<A`9,<$8,$9A$EA9A8),L$$
$$$$$$$$$$
"L$X+G;I)<G)8;<,$;+$J)`6;<,$FB<()*+,$$
FY,<($9A$XKC$9A$=9+<$>6&*+,'(,$8A'<$9A$=;=+9A();'$GA+GA<),'',$D$/.-000$'A)<<A'G,<Z$/.--$
=;6(,+6<$3&(&6;MB?;(,<L$$
c&>)')();'$D$FY,<($+',$=A(3;9;?),$8,<$<&G6&();'<$,N;G6)',<Z$A';6EA9,E,'($&=A)<<,<$,($P$
,N=6,<<);'$IA6)A`9,L$
a)?',<$G9)')*+,<$D$$
4 :L7<I:57>?&@8AL7@4<B7@8&C!5)*#!(80D&E!(*,.'+&05.)'(!>,)'0D)F9*1%)'$.,&($$
4 GB;C<7B;&6798A<7D8&D$$
=A'G6&A<$O-.$,,D6&!0D,)'.2*&E!DG9)5,)9D.&!E!-6'*5,.5.)'$Q$
>;),Z$h:$O$.058,&!E!0.,,D)(&!>.1.$.,&$Q$$

)'(,<()'<$O.1&*(!%60)'.$1!E!9,)1$9(*(!,&05$1Q$$
4 <@4C<>A&9:;7<45&D$(56,.1.56E!DG9)+&,5.1.56!:HH%$(0*1.'&=$
4 954;68A&A>6B@7L4@8A&)&$')%$1.&(!-&!1$!(*&*,I&
a+<=)G);'$,($8)A?';<()G$D$$
i9$>A+($A>AL8C<8@$G,((,$EA9A8),$D$
4 ,'$;:B;4<45&D$9,<$<)?',<$=6&<,'(<$8A'<$/0j$8,<$GA<$$<;'($9,<$;`<(6+G();'<$
)'(,<()'A9,<Z$<)?',<$8Y+'$)9,+<$E&G;')A9$O=A<$8,$9)`&6A();'$8,$<,99,<$8A'<$9,<$f23Q$
4 G3,M$58A&8;G4;<A$D$9,<$<)?',<$<;'($+'$6,(A68$8,$G6;)<<A'G,$,($8,<$)'>,G();'<$
6,<=)6A(;)6,<L$$
4 G3,M$58A&46>5<8A&D$<(&6)9)(&$
U,$8)A?';<()G$,<($+'$(,<($`);G3)E)*+,$D$&9&IA();'$8,$!5&64;A&54&A>8>@$g$P$b0$EE;9.U$$
Ok$(,<($8,$9A$<+,+6$lQZ$A=6H<$<,+9,E,'($/$E;)<$G3,M$mmL$$
"I;9+();'$D$<&IH6,$G3,M$=9+<$8,$#0j$*+)$EH','($P$8,<$8&GH<$AIA'($-0$A'<L$
@6A)(,E,'($D$)9$6,=;<,$<+6$$=6)<,$,'$G3A6?,$=6&G;G,$6,<=)6A(;)6,Z$8)?,<()I,Z$I;)6,$+',$
(6A'<=9A'(A();'$=+9E;'A)6,$,(.;+$3&=A()*+,L$$
FA+<,<$D$$
U,<$GA+<,<$8,$G,((,$XKC$<;'($8,<$E+(A();'<$,'$n*1/$<+6$9,$?H',$G;8A'($=;+6$$
FJ@C$OGB<()G$>)`6;<)<$(6A'<E,E`6A',$G;'8+G(A'G,$6,?+9A(;6QZ$FJ@C$&(A'($+'$6&?+9A(,+6$
8,$9A$=,6E&A`)9)(&$8+$GA'A9$F9L$
! ';E`6,+<,<$E+(A();'<$O-00Q$4g$;'$';(,$+',$D656,)?6'6.56!$1161.2*&$O)9$,N)<(,$
=9+<),+6<$E+(A();'<$=;<<)`9,$<+6$G,$?H',$,($,'(6A)'A'($9A$X+G;I)<G)8;<,QL$cA'<$
=;=+9A();'$GA+GA<),'',Z$9A$E+(A();'$9A$=9+<$>6&*+,'(,$,<($9A$8&9&();'$8,<$(6;)<$
G;8;'<$G;8A'($=;+6$5d"$,'$=;<)();'$oJ#02L$$
J)*,2*).!0&55&!-6165.)'!&(5!(.!+,62*&'5&!-$'(!1$!9)9*1$5.)'!0$*0$(.&''&!&5!9)*,2*).!.1!G!$!
$*5$'5!-4D656,)KG?)5&(!:L/MM=!N$i9$',$>A+($=A<$;+`9),6$9Y)'>9+,'G,$8,$5U7;<8@4C<7B;&8;<@8&
8;D7@B;;8?8;<&8<&9:;:<7=>8$D$9Y3&(&6;MB?;(),$<,6A)($+'$AIA'(A?,$A+$<,'<$?&'&()*+,$GA6$
GY,<($+',$E+(A();'$*+)$G;'>H6,$+',$6&<)<(A'G,$AGG6+,$A+N$)'>,G();'<$)'(,<()'A9,<$G;EE,$
9,$F3;9&6AZ$?6\G,$A+N$<&G6&();'<$=9+<$&=A)<<,<$6,'G;'(6&,<$=;+6$G,$=3&';(B=,L$F,((,$
6&<)<(A'G,$A<<+6,$+',$E,)99,+6,$<+6I),$8,<$=;=+9A();'<$>AG,$P$G,<$)'>,G();'<$,($9YA99H9,$
<Y,<($A)'<)$6&=A'8+L$
c&=)<(A?,$'&;'A(A9$D$
U,$8&=)<(A?,$'&;'A(A9$,<($+'$(,<($`);G3)E)*+,$>)A`9,$,($$=,6E,($+',$=6)<,$,'$G3A6?,$
=6&G;G,$D$9,$(,<($8,$%+)(6B$,<($<B<(&EA()*+,$,'$J6A'G,L$i9$G;'<)<(,Z$P$1$R;+6<$8,$I),Z$,'$
8&=;<,6$+'$?;+((,$8,$<A'?$8,<$,'>A'(<$<+6$+'$=,()($<=;($<+6$+'$=A=),6$`+IA68$,($;'$8;<,$
+',$,'MBE,$=A'G6&A()*+,Z$54&0@KLA7;8&.??>;B@:4C<7C8&E0.$F&D$+',$@iC$A+?E,'(&,$,<($
9,$<)?',$8Y+',$8&>A)99A'G,$=A'G6&A()*+,$,($$)9$A$&(&$;`<,6I&$*+Y)9$B$AIA)($+',$G;66&9A();'$
,'(6,$9,$(A+N$8,$@iC$8A'<$9,$<A'?$,($9A$=6;`A`)9)(&$8YW(6,$=;6(,+6$8Y+',$E+(A();'$FJ@CL$$$
4 <)$9A$@iC$_$<,+)9$D$9,$8&=)<(A?,$,<($'&?A()>$
4 <)$9A$@iC$g$<,+)9$D$9,$8&=)<(A?,$,<($=;<)()>$,($;'$&(+8),$,'<+)(,$9,$?H',$G;8A'($=;+6$
FJ@CL$7'$A'A9B<,$=6&G)<&E,'($9,<$-p$E+(A();'<$8,$G,$?H',$FJ@C$*+)$
G;66,<=;'8,'($P$p2j$8,<$E+(A();'<$8A'<$9A$=;=+9A();'$GA+GA<),'',L$a)$9,$
9A`;6A(;)6,$8,$`);9;?),$E;9&G+9A)6,$(6;+I,$*+,$9Y)'8)I)8+$,<($<;)($3;E;MB?;(,$<;)($
3&(&6;MB?;(,$=;+6$+',$E+(A();'Z$)9$IA$>A)6,$=A6$<&G+6)(&$+'$(,<($8,$9A$<+,+6L$a)$;'$
'YA$=A<$(6;+I&$8,$E+(A();'$<+6$9,<$-$A99H9,<Z$;'$6,>A)($9,$8;<A?,$8,$9A$@iC$A+$-/HE,$
R;+6$G3,M$9Y,'>A'($D$<)$G,$G;'(6q9,$,<($)'>&6),+6$A+$<,+)9Z$;'$A66W(,$(;+(,$6,G3,6G3,Z$
,'$6,IA'G3,$<Y)9$,<($<+=&6),+6$;'$>A)($9,$(,<($8,$9A$<+,+6L$$

$
XA93,+6,+<,E,'($;'$',$=,+($=A<$>A)6,$9,$(,<($8,$9A$<+,+6$AIA'($/$E;)<Z$GY,<($
=;+6*+;)$;'$6&A9)<,$9,<$A+(6,<$(,<(<$AIA'(L$F,$(,<($8,$9A$<+,+6$<,$>A)($8A'<$9,<$
O&'5,&(!-&!B&(()*,0&(!!&5!-&!O)%965&'0&(!9)*,!1&!@*0)<.(0.-)(&$D$$
4 <Y)9$,<($A';6EA9Z$;'$G;'<)8H6,$*+,$9Y)'8)I)8+$,<($A((,)'($,($;'$=6,'8$,'$G3A6?,$9A$
EA9A8),$$
4 <Y)9$,<($';6EA9Z$;'$G;'<)8H6,$*+,$9Y)'8)I)8+$,<($3&(&6;MB?;(,$,($;'$IA$8)6)?,6$9,<$
=A6,'(<$I,6<$9,$G;'<,)9$?&'&()*+,L$
$
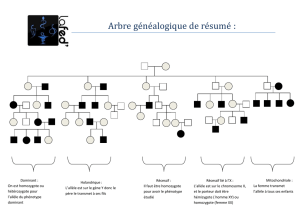
F;'<,)9$%&'&()*+,$D$)9$,<($)99+<(6&$P$9YA)8,$8,$f$GA<L$$$
FA<$'/$$
U,$=6,E),6$GA<$<,$=6&<,'(,$8,$G,((,$>AV;'$D$
+'$G;+=9,$I),'($I;+<$I;)6$,($I;+<$8,EA'8,$
9,$6)<*+,$*+,$9,+6$,'>A'($<;)($A((,)'($8,$
E+G;I)<G)8;<,$<AG3A'($*+,$9A$EH6,$$8,$G,$
G;+=9,$A$+',$<r+6$*+)$A$+',$>)99,$EA9A8,L$
7'$G;EE,'G,$=A6$8)6,$*+,$9A$<r+6$,<($
3&(&6;MB?;(,$;`9)?A(;)6,E,'($$8;'G$+'$8,$
<,<$8,+N$=A6,'(<$,<($(6A'<E,((,+6L$U,$
6)<*+,$=;+6$9A$EH6,$*+)$I),'($8,$9A$
=;=+9A();'$?&'&6A9,$A$8;'G$+'$6)<*+,$
8YW(6,$3&(&6;MB?;(,$P$/.-0$OA66;'8)$8,$
/.--QL$XA)'(,'A'($;'$G;''As($9,$6)<*+,$*+,$
9,<$=A6,'(<$<;),'($3&(&6;MB?;(,<Z$,($
G;EE,$;'$<A)($*+,$8,<$=A6,'(<$3&(&6;MB?;(,<$;'($-#j$8,$6)<*+,$8YAI;)6$+'$,'>A'($
EA9A8,Z$A9;6<$;'$,'$8&8+)($9A$9,)>$>.1.56!2*&!1&*,!&'+$'5!().5!$55&.'5!P!L/M#L/MQ#L/R!P!
L/SQ!O/.-000$8A'<$9A$=;=+9A();'$?&'&6A9,QL$U,$G;'<,)9$?&'&()*+,$IA$8&(,6E)',6$9A$
E+(A();'$G3,M$9A$<r+6L$a)$9A$EH6,$'Y,<($=A<$=;6(,+<,$8,$9A$E+(A();'Z$;'$'YA'A9B<,$=A<$9,$
?&';E,$8+$=H6,L$"'$6,IA'G3,$<)$9A$EH6,$,<($=;6(,+<,$8Y+',$E+(A();'Z$;'$IA$A'A9B<,6$9,$
?H',$FJ@C$=;+6$9,<$-p$E+(A();'<$9,<$=9+<$>6&*+,'(,<$8A'<$9A$=;=+9A();'$?&'&6A9,$8,$
EA')H6,$P$8)E)'+,6$A+$EAN)E+E$9,+6$6)<*+,L$aY)9$'Y,<($=A<$=;6(,+6$8,$G,<$E+(A();'<Z$
9,+6$6)<*+,$8YAI;)6$+'$,'>A'($A((,)'($6,8,<G,'8$P$G,9+)$8,$9A$=;=+9A();'$?&'&6A9,L$a)$=A6$
MUCOVISCIDOSE
Conseil génétique
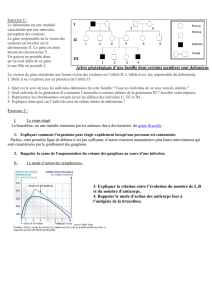
Parents d ’un enfant atteint ont un risque 1/4 d ’avoir un enfant atteint
risque pour la fratrie d’un parent atteint?
Probabilité que II-3 soit hétérozygote = 1/2
Probabilité que II-4 soit hétérozygote = 1/20
Probabilité que III-3 soit atteint = 1/2 X 1/20 X 1/4
=
1/160
I
II
III ?
4
3
123
alors que couple population générale 1/2 000
Dépistage des hétérozygotes
DEPISTAGE NEONATAL de la Mucoviscidose
Dosage TIR-J3 Dosage TIR-J3 < seuil
STOP
Dosage TIR-J3 > seuil
DEPISTAGE
Etude du gène CFTR
M/M M/X X/X
Control TIR-J21 < seuil
Dosage TIR-J21
Dosage TIR-J21 > seuil
Test de la sueur
Anormal Normal
Prise en
Charge
maladie
Conseil
génétique
si M/X
BIOLOGIE
MOLECULAIRE
CRCM
Papier buvard dosage de la trypsine immunoréactive (TIR) (enzyme pancréatique)
Centre de Ressources
et de Compétences
pour la Mucovisidose
29 mutations
Sensibilité 98%
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%