La ou les résistances à l’aspirine ? m i

159
Correspondances en Risque CardioVasculaire - Vol. II - n° 4 - octobre-novembre-décembre 2004
La ou les résistances
à l’aspirine ?
■
■
L. Drouet*
L’
aspirine (figure 1) est une thérapeu-
tique que nous utilisons depuis la
plus haute antiquité, d’abord comme
anti-inflammatoire, puis, en plus, comme anti-
thrombotique. On pourrait imaginer que, sur
une molécule aussi ancienne, on sache tout et
qu’on n’ait plus rien à dire. Et bien non, nous
découvrons régulièrement des fonctions, nous
affinons nos connaissances sur les effets béné-
fiques et les effets délétères. Et ce dans des
contextes de modes et de débats d’actualité.
Pour une molécule aussi vieille, c’est un
comble ! Le dernier débat d’actualité (avant
celui de l’effet potentiellement prothrombo-
tique des inhibiteurs spécifiques de COX-2,
débat qui n’est pas très éloigné de ceux tou-
chant l’aspirine) est celui qui se poursuit autour
du concept des résistances à l’aspirine.
Pourtant, ce concept est ancien, probablement
aussi ancien que l’utilisation de l’aspirine
comme thérapeutique interférant sur les sys-
tèmes de l’hémostase. L’apparition de l’aspirine
comme thérapeutique antithrombotique pla-
quettaire a permis de démontrer que, dans
toutes les grandes méta-analyses de patients
atteints des différentes manifestations de la
pathologie athérothrombotique, ce traitement
prévenait la récidive des accidents thrombo-
tiques artériels dans environ 25 à 30 % des cas.
Ce qui veut dire que 70 à 75 % des survenues ou
des récidives des accidents thrombotiques de
ces patients présentant une pathologie athéro-
thrombotique et traités par l’aspirine ne sont
pas prévenues. Dans ces conditions, la résistan-
ce à l’aspirine ne peut en aucun cas être fondée
sur une définition clinique. En effet, la survenue
mise au point
* Service d’angio-hématologie biologique,
hôpital Lariboisière, Paris.
■
■ Les grands essais thérapeutiques nous ont appris que l’aspirine ne prévenait que
25 à 30 % des accidents ischémiques cardiovasculaires. Ce qui veut dire que 70 à
75 % des récidives échappent à cette prévention.
■
■ Les plaquettes ne sont pas les seules cellules à synthétiser les thromboxanes ; par
exemple, l’augmentation de production de thromboxanes au moment des acci-
dents ischémiques est établie de longue date.
■
■ La cause la plus fréquente de résistance à l’aspirine est une non-observance du
traitement.
■
■ La cause la plus fréquente de l’inefficacité de l’aspirine est un problème de dose
insuffisante.
■
■ Les autres causes sont principalement :
– ∑ l’interférence médicamenteuse,
–∑ des mutations des gènes cibles.
■
■ Il est nécessaire de confirmer, par de larges essais cliniques bien conduits, que la
résistance biologique a une signification clinique, de sorte que soit attesté l’intérêt
qu’il y a à la rechercher et à la traiter.
... Points forts ...
... Points forts ...
H
H
H
H
H
O
O
O
O
CC
C
CC
C
C
C
C
H
H
H
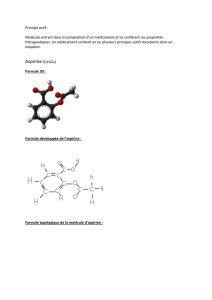
Figure 1. Formule chimique de l’aspirine.

Correspondances en Risque CardioVasculaire - Vol. II - n° 4 - octobre-novembre-décembre 2004
160
ou la récidive d’un accident thrombotique arté-
riel chez un patient traité par aspirine peut pro-
bablement être due au fait que l’accident isché-
mique n’est pas d’origine thrombotique, et donc
qu’il n’est pas prévenu par une thérapeutique
antiplaquettaire. Même si l’accident ischémique
est d’origine thrombotique, sa survenue ou sa
récidive est de participation mixte (plaquettes et
coagulation) et, dans ces conditions, une théra-
peutique antiplaquettaire stricte n’est pas suffi-
sante pour prévenir l’accident thrombotique. Et
même si sa survenue ou sa récidive est d’origine
principalement plaquettaire, la voie des prosta-
glandines plaquettaires inhibées par l’aspirine
n’est qu’une des nombreuses voies de recrute-
ment et d’activation plaquettaire et, dans ces
conditions, une efficacité, même totale, de l’as-
pirine n’est pas suffisante pour inhiber globale-
ment la fonctionnalité plaquettaire et mettre à
l’abri de tout développement thrombotique pla-
quettaire. Si l’on s’accorde à prendre comme
définition de la résistance à l’aspirine le fait que
celle-ci n’exerce pas l’effet pharmacodynamique
attendu sur les plaquettes, ce sont nos collègues
neurologues qui les premiers ont utilisé le terme
de résistance (et d’échappement) à l’aspirine, et
ce il y a plus de 10 ans. Cette résistance est indis-
sociable du problème de “la bonne dose d’aspi-
rine”, une autre question qui a agité pendant la
ou les deux décennies précédentes le microcos-
me des utilisateurs de l’aspirine. Les stratégies
d’utilisation, en prévention cardiovasculaire, de
doses de plus en plus faibles d’aspirine (sans
diminution significative de l’efficacité dans les
grands essais randomisés) amènent à une évi-
dence qui relève du bon sens médico-scienti-
fique mais qui entre en contradiction avec ce que
semblent vouloir dire les grandes méta-ana-
lyses : plus on diminue la dose, plus on a de
risques de voir apparaître au niveau individuel
des patients pour lesquels la dose sera insuffi-
sante. Il s’agit donc d’un problème quotidien.
R
ÉSISTANCE À L
’
ASPIRINE
:
LES ENTITÉS CLINICO
-
BIOLOGIQUES
Ce terme est parfois employé en cas de récidives
d’accidents ischémiques, intervenant malgré un
traitement apparemment bien conduit, et lors-
qu’une inefficacité clinique est observée. Les
grands essais thérapeutiques nous ont appris
que l’aspirine ne prévenait que 25 à 30 % des
accidents ischémiques cardiovasculaires ; ce qui
veut dire que l’on sait que 70 à 75 % des réci-
dives échappent à cette prévention. Cette récidive
clinique a plusieurs types d’explications :
➊Récidive clinique d’un accident non isché-
mique.
➋Récidive clinique d’un accident ischémique mais
de physiopathologie autre que thrombotique.
➌Récidive clinique d’un accident ischémique
thrombotique mais de physiopathologie autre
que thromboplaquettaire.
➍Récidive clinique d’un accident ischémique
thrombotique de physiopathologie thrombopla-
quettaire, physiopathologie pour laquelle l’inhi-
bition de la voie des prostaglandines plaquet-
taires est insuffisante au regard de l’efficacité
attendue. Cette dernière peut avoir deux types
d’explications :
• Des prostaglandines, d’une origine autre que
plaquettaire, viennent induire l’activation des
plaquettes (même si leur propre voie endogène
des prostaglandines est correctement inhibée
par l’aspirine). Pour comprendre ce type de
résistance, il faut se rappeler que les plaquettes
ne sont pas les seules cellules à synthétiser les
thromboxanes. De nombreuses autres cellules
peuvent les synthétiser, et en particulier
d’autres cellules vasculaires intervenant dans la
pathologie thrombotique, principalement les
monocytes et les cellules musculaires lisses. La
voie de synthèse des thromboxanes dans ces
cellules ne passe pas par la COX-1 constitution-
nelle mais par la voie de la COX-2 inductible. Or,
la COX-2 est beaucoup moins sensible à l’acéty-
lation par l’aspirine que la COX-1.
La majorité des accidents thrombotiques de
l’athérosclérose sont la conséquence de la rup-
ture (ou de l’érosion) d’une plaque d’athéro-
sclérose (accidents ischémiques d’origine athé-
rothrombotique). Les mécanismes de rupture
font intervenir, en particulier, les médiateurs de
l’inflammation et les monocytes. L’activation
locale des monocytes (réaction inflammatoire)
joue un rôle pivot dans les phénomènes d’insta-
bilité, comme en témoigne la forte valeur pré-
mise au point
Quand on suit la littérature
au sujet de la résistance à
l’aspirine, on ne peut que
constater que se trouvent
réunies sous le terme de
“résistance à l’aspirine” plu-
sieurs entités complètement
différentes ; tout le monde ne
parle donc pas du tout de la
même chose.

161
Correspondances en Risque CardioVasculaire - Vol. II - n° 4 - octobre-novembre-décembre 2004
dictive des marqueurs/agents d'inflammation
(protéine C réactive [CRP], interleukine 6) sur le
risque ischémique cardiovasculaire. Cette acti-
vation aboutit à une synthèse extraplaquettaire
augmentée de thromboxanes capable d’activer
les plaquettes (même les plaquettes dont la
voie endogène des prostaglandines a été cor-
rectement inhibée par l’aspirine). L’augmen-
tation de production de thromboxanes au
moment des accidents ischémiques est établie
de longue date. Un travail récent, qui a repris un
sous-groupe des 5 529 patients canadiens de
l’étude HOPE, recevant tous de l’aspirine (dose
non précisée), a pu mesurer les thromboxanes
urinaires (recueil d’urines à l’inclusion dans
l’étude) chez les 488 patients ayant eu, au cours
de la période de suivi, un accident cardiovascu-
laire grave (ceux-ci étant comparés à 488 pa-
tients appariés mais n’ayant pas eu d’accident).
Les patients se trouvant dans le quartile supé-
rieur du taux de thromboxanes urinaires (qui ont
un taux à plus de deux fois celui du quartile infé-
rieur) ont un risque 1,8 fois plus important
d’avoir un accident cardiovasculaire.
Cette physiopathologie ne permet pas d’allé-
guer, à proprement parler, d’une résistance des
plaquettes à l’aspirine, puisque les throm-
boxanes extraplaquettaires non inhibés par de
faibles doses d’aspirine conservent un pouvoir
activant des plaquettes “aspirinées” à dose effi-
cacement antiplaquettaire.
• Une autre explication est conceptuellement
proche de la précédente mais utilise des intermé-
diaires différents. Les modifications oxydatives de
l’acide arachidonique ne se limitent pas à la for-
mation enzymatique d’éicosanoïdes. L’acide ara-
chidonique peut être attaqué par les radicaux
libres et donner naissance aux isoprostanes. Ces
isoprostanes peuvent, entre autres, activer les pla-
quettes par leurs récepteurs aux thromboxanes.
Une production excessive d’isoprostanes est asso-
ciée aux facteurs de risque cardiovasculaire
majeurs (tabac, hypertension artérielle, diabète,
hypercholestérolémie). Du fait de l’origine non
enzymatique des isoprostanes, l’aspirine n’a pas
d’effet sur leur production. Une production accrue
de ces métabolites pourrait donc expliquer la sur-
venue d’accidents ischémiques impliquant les pla-
quettes dans les pathologies cardiovasculaires
malgré un traitement par aspirine bien conduit ;
mais, là encore, il ne s’agit pas à proprement par-
ler de “résistance à l’aspirine” (figure 2).
• Utilisation d’autres voies alternatives d’activa-
tion et de recrutement plaquettaire (voie de
l’ADP, voie des catécholamines, voie de la séro-
tonine, etc.).
➎Accident ischémique thrombotique de physio-
pathologie thromboplaquettaire pour laquelle
l’inhibition de la voie des prostaglandines aurait
pu être efficace, mais où la dose administrée (ou
le mode d’inhibition) est insuffisante pour obte-
nir l’inhibition efficace de la voie des prosta-
glandines plaquettaires.
➏Accident ischémique thrombotique de phy-
siopathologie thromboplaquettaire sur laquelle
l’inhibition de la voie des prostaglandines aurait
pu être efficace, mais où l’aspirine (quelle que
soit la dose administrée) ne permet pas d’obte-
nir l’inhibition requise de la voie des prostaglan-
dines plaquettaires.
Pour déterminer si les plaquettes subissent effi-
cacement l’effet inhibiteur de l’aspirine sur la
synthèse des prostaglandines, il faut disposer
de tests pharmacodynamiques permettant de
mesurer précisément et spécifiquement cet
effet de l’aspirine sur les plaquettes. On possède
deux types de tests :
– un test fonctionnel de mesure de l’agrégation
plaquettaire spécifiquement initiée par de
faibles doses d’acide arachidonique ;
Figure 2. Effet de l’aspirine sur la voie de l’acide arachidonique.
Phospholipides membranaires
Acide arachidonique
Phospholipases
Cyclo-oxygénase de type 1
Aspirine
Endoperoxydes
Thromboxane A2
Agrégation plaquettaire
Thromboxane synthase

Correspondances en Risque CardioVasculaire - Vol. II - n° 4 - octobre-novembre-décembre 2004
162
– ou encore, plus spécifiquement : un test bio-
chimique de mesure de la quantité de throm-
boxanes formée par les plaquettes après activa-
tion. Le moyen habituel est de mesurer le throm-
boxane A2– ou plutôt son métabolite stable non
enzymatique, le thromboxane B2– dans un
sérum produit dans des conditions standardi-
sées. En effet, au cours de la coagulation du
sang, les plaquettes sont activées et s’agrègent
entre elles et avec le réseau de fibrine. Le caillot
fibrinoplaquettaire, en se rétractant, libère le
sérum, qui contient donc les produits de sécré-
tion et de synthèse des plaquettes (en particu-
lier les thromboxanes).
Si l’effet de l’aspirine est efficace, l’agrégation à
l’acide arachidonique est totalement abolie
(dans le test fonctionnel) et les plaquettes ne
peuvent plus produire de thromboxanes (dans
le test biochimique).
L’inhibition de l’agrégation plaquettaire induite
par l’acide arachidonique (test dans lequel les
voies d’activation plaquettaire ne sont activées
que par la seule voie des prostaglandines) ne
doit pas être confondue avec les tests globaux
d’évaluation des fonctions plaquettaires voire
de tout ou partie de l’hémostase primaire. La
persistance d’un certain degré de réactivité pla-
quettaire malgré le traitement par aspirine ne
signifie pas obligatoirement que la voie des
prostaglandines plaquettaires n’est pas totale-
ment inhibée.
On comprend donc que la notion de résistance
biologique elle-même va totalement dépendre du
test d’évaluation des plaquettes. Ce n’est pas
toujours facile pour le non-spécialiste de s’y
retrouver, car les tests sont nombreux. De nou-
veaux tests apparaissent régulièrement, et les
fabricants concepteurs peuvent les présenter
comme spécifiques même s’ils ne le sont pas
totalement. Ces tests évaluent une ou plusieurs
fonctions plaquettaires qui sont plus ou moins
sensibles à la voie des prostaglandines inhibée
par l’aspirine, et ils sont donc plus ou moins
représentatifs de la pathologie humaine. On voit
par conséquent que l’on peut avoir une très large
différence dans les résultats en fonction du test
choisi et du degré de persistance d’une réactivité
plaquettaire considérée comme une résistance.
La non-inhibition attendue peut alors être
considérée comme une résistance ou une non-
activité. Cette résistance est due le plus sou-
vent à une non-observance du traitement.
L’aspirine est une thérapeutique “banale” : le
patient n’a pas l’impression qu’en prenant de
l’aspirine il prend une thérapeutique majeure.
Elle peut aussi être due au fait que l’aspirine
ne produit pas l’effet attendu. Il y a plusieurs
explications potentielles à cette non-efficacité
(ou à cet échappement, c’est-à-dire lorsqu’une
dose efficace devient inefficace). La plus fré-
quente peut être un problème de dose. Si
l’augmentation de la dose fait apparaître (ou
réapparaître) l’efficacité pharmacodynamique,
il ne s’agit pas d’une résistance “vraie” mais
plutôt d’un problème de sensibilité ou de
dosage. Une des explications à cette nécessité
d’une dose plus forte réside dans l’existence,
chez le patient à traiter, d’un chiffre de pla-
quettes, d’un
turn-over
ou d’une activité des
plaquettes augmentés.
Mais d’autres explications doivent aussi être
envisagées dans le cas d’une inefficacité d’un
traitement par l’aspirine réellement absorbé, et
premier lieu une interférence médicamenteuse.
Le risque d’interférence négative de certains
AINS sur l’effet de l’aspirine sur les plaquettes
(figure 3) a été mis en évidence par le groupe de
G.A. FitzGerald. Celui-ci a en effet montré que
l’administration d’AINS (et en particulier d’ibu-
profène) avant la prise quotidienne d’aspirine
rendait temporairement le site sensible de la
COX-1 inaccessible à l’acétylation par l’aspirine.
Si bien que, lorsque l’AINS se détache de la
COX-1, son effet inhibiteur disparaît sans que
l’effet antiplaquettaire de longue durée d’action
de l’aspirine ait pu se mettre en place. Depuis
cette étude d’interférence pharmacodyna-
mique, une étude épidémiologique est venue
conforter cette démonstration pharmacologique
en montrant que tous les critères de jugement
cardiovasculaires, dont la mortalité globale,
sont accrus chez les patients cardiovasculaires
traités par aspirine et recevant par ailleurs de
l’ibuprofène, par rapport aux patients ne rece-
vant que de l’aspirine.
Dans ce type de considérations, le patient n’est
pas à proprement parler résistant à l’aspirine,
mise au point
Figure 3. Plaquettes sanguines.

163
Correspondances en Risque CardioVasculaire - Vol. II - n° 4 - octobre-novembre-décembre 2004
mais la comédication fait perdre, au moins en
partie, le bénéfice du traitement par l’aspirine.
À coté de ces causes admises et démontrées de
résistance à l’aspirine, plusieurs hypothèses
additionnelles peuvent être envisagées.
Les mutations génétiques des cibles
La cible de l’aspirine est, comme précédemment
expliqué, le site actif de la COX-1, qui va être
acétylé par l’aspirine. Il est alors envisageable
que des mutations sur ou à proximité de ce site
puissent gêner l’action de l’aspirine. D’autres
hypothèses de mutations de voies métabo-
liques d’activation ou d’inhibition pourraient
aussi expliquer que l’aspirine puisse être finale-
ment inactive. Parmi les effecteurs plaquet-
taires, un lien entre le polymorphisme PLA2 de
la glycoprotéine GP IIb/IIIa de la membrane pla-
quettaire et la résistance à l’aspirine a aussi été
rapporté.
La notion de résistance à l’aspirine amène donc
à soulever deux points principaux :
• Le premier est celui de la nécessité d’un
consensus sur la définition de la résistance et
sur le test à utiliser pour la reconnaître, de
manière à ce que tous ceux qui en parlent évo-
quent le même phénomène.
• Le deuxième repose sur l’importance qu’il y a
à confirmer, par de larges essais cliniques bien
conduits, que la résistance biologique ainsi défi-
nie a une signification clinique, ce qui prouverait
l’intérêt qu’il y aurait à la rechercher et à la trai-
ter (au mieux préventivement).
Elle pose également des questions pratiques
majeures :
• S’agit-il d’une pathologie ne relevant pas d’un
phénomène thromboplaquettaire, de sorte que
les antiplaquettaires ne seraient pas la bonne
cible pour obtenir l’efficacité ?
• S’agit-il d’une “vraie” résistance à l’aspirine et
peut-on la contourner, en particulier en adminis-
trant une dose individuellement efficace ?
• S’agit-il d’une pathologie thromboplaquettaire
dans la pathogénie de laquelle la voie des pros-
taglandines ne joue pas le rôle principal et pour
laquelle une autre cible, seule ou en associa-
tion, peut être préconisée ?
Trente ans après les premières discussions sur
la bonne dose d’aspirine, la question s’est sin-
gulièrement compliquée, avec une accumula-
tion de données nouvelles. Mais, en pratique,
elle reste posée, et le restera tant qu’une défini-
tion claire et une attitude biologico-clinique
cohérente ne seront pas proposées et adoptées
par la communauté médicale.
Considérer les antiplaquettaires, et en particu-
lier l’aspirine, comme une thérapeutique qui
n’aurait pas besoin de monitoring est peut-être
finalement un tort. La question peut se poser de
savoir si le test de monitoring le plus répandu
qu’est l’agrégation plaquettaire ne va pas deve-
nir un test de routine (peut-être sous une forme
simplifiée ou au lit du patient).
P
OUR EN SAVOIR PLUS
...
• Christiaens L, Macchi L. Aspirin resistance 2003: a review
of the literature. Arch Mal Cœur Vaiss 2004;97(4):320-6.
• Patrono C. Aspirin resistance: definition, mechanisms and
clinical read-outs. J Thromb Haemost 2003;1(8):1710-3.
• Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and
the issue of drug resistance. Arterioscler Thromb Vasc Biol
2004;24(11):1980-7.
• Hennekens CH, Schror K, Weisman S, Fitzgerald GA. Terms
and conditions: semantic complexity and aspirin resistance.
Circulation 2004;110(12):1706-8.
1
/
5
100%