Cinétique enzymatique Mesure de l’activité enzymatiqaue

Cinétique enzymatique
Mesure de l’activité
enzymatiqaue
Dr AKSAS
1

Cinétique enzymatique
La R enzymatique peut être subdivisée en 3
étapes:
1. Phase préstationnaire;
2. Phase stationnaire;
3. Phase post-stationnaire.
2

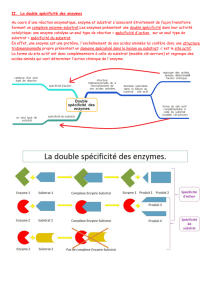
Cinétique Michaelienne
Représentons les vitesses initiales de R en
fonction des concentrations du substrats
3

Cinétique Michaelienne
Les vitesses mesurées croissent en fonction des
concentrations du substrat.
Mais cette relation n’est pas linéaire : le graphe de
cette fonction est une hyperbole;
Lorsque la concentration du substrat est nulle la
vitesse est évidemment 0, donc l’hyperbole passe
par l’origine du graphe.
4

5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
1
/
64
100%