4-Douleur postopératoire.indd

COMMENT UTILISER LES
ANALGÉSIQUES DE PALIER 2
DANS LE CADRE DE LA
DOULEUR POSTOPÉRATOIRE ?
Marcel Chauvin
Service d’Anesthésie Réanimation, Hôpital Ambroise Paré, 9 avenue
Charles de Gaulle, 92100 Boulogne
INTRODUCTION
Les analgésiques de palier 2 de la classification OMS regroupent des médica-
ments de classes différentes, dont la puissance d’action est intermédiaire entre
celle des morphiniques puissants comme les morphinomimétiques (morphine,
fentanyl et dérivés, péthidine, méthadone…) et celle des analgésiques comme le
paracétamol et les AINS. En fait, cette classification est quelque peu artificielle car
certaines substances du palier 2 peuvent être autant, voire moins efficaces que
celles du palier 1 comme les AINS et inversement certains analgésiques du palier
2 peuvent générer une analgésie équivalente avec autant d’effets indésirables
que les analgésiques du palier 3. Par ailleurs, codéine et dextropropoxyphène
sont des morphinomimétiques qui créent autant de dépression respiratoire que
la morphine à dose équipotente.
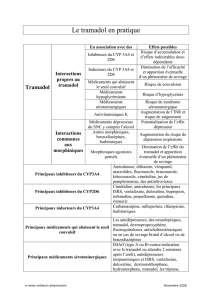
1. TRAMADOL
Le tramadol est un analgésique central de synthèse dont le profil d’action
très particulier et original combine plusieurs mécanismes qui agissent sur des
cibles complémentaires : une activité morphinique faible, 6 000 fois plus faible
que celle de la morphine, et une activité mono-aminergique qui consiste en
une inhibition des recaptures de la noradrénaline et de la sérotonine. En fait,
c’est le dérivé O-déméthylé (M1), 200 fois plus affine pour les récepteurs µ que
la molécule mère, qui apporte la contribution majeure de l’action morphinique
du tramadol [1]. Les actions morphiniques et non morphiniques du tramadol
agissent de manière synergique sur les voies descendantes inhibitrices du
SNC, avec pour corollaire une modulation des neurones de deuxième ordre au
niveau spinal. L’origine de ces voies descendantes se situent au niveau du tronc
cérébral (noyaux du raphé, substance grise périacqueducale, locus coeruleus et
projections reticulo-spinales).

MAPAR 2006
148
Le tramadol et ses métabolites sont éliminés en majorité au niveau rénal. Les
patients ayant une insuffisance rénale ont une réduction de l’excrétion de trama-
dol et de son métabolite M1. En cas d’insuffisance hépato-cellulaire, la clairance
métabolique du tramadol est diminuée et la demi-vie terminale est doublée. Ainsi
il est conseillé de diminuer de moitié la dose de tramadol administrée en cas
d’insuffisance rénale ou d’insuffisance hépatique. En cas d’insuffisance rénale
majeure ou d’anurie, l’accumulation de M1 peut être à l’origine de dépression
respiratoire avec apnée, contre-indiquant son utilisation.
La biodisponibilité du tramadol par voie orale est élevée, 70 à 90 %, et
approche les 100 % après des prises multiples, probablement par saturation du
métabolisme de premier passage hépatique. Ainsi, la dose recommandée de
tramadol par voie orale est proche de celle de la voie IV.
Les effets indésirables du tramadol ressemblent à ceux des opiacés à
l’exception de la dépression de la fonction respiratoire qui est absente aux
doses thérapeutiques. Ils sont identiques en nature pour les voies parentérale
et orale, cependant ils sont globalement moins fréquents pour la voie orale
qu’intraveineuse et pour les formes à libération prolongée qu’immédiate [2]. Les
effets les plus fréquents sont les nausées, vomissements, vertiges, céphalées
et sécheresses de bouche. Les nausées et vomissements sont plus fréquents
avec des administrations en bolus qu’en perfusion continue et sont dose-dépen-
dants. L’ondansétron n’est pas efficace pour prévenir ou traiter les nausées et
vomissements du tramadol et antagonise partiellement l’effet analgésique du
tramadol probablement par ses propriétés antagonistes 5HT3 [3, 4]. Par contre,
le dropéridol à doses faibles (bolus de 1,25 mg) est particulièrement efficace et il
est conseillé d’associer du dropéridol (50 µg.ml-1) dans la PCA de tramadol [5].
Un autre moyen pour réduire l’incidence des nausées et vomissements du
tramadol consiste à administrer, en peropératoire, la dose de charge (100 mg)
qui précède la perfusion continue. Ceci a par ailleurs l’intérêt d’anticiper le pic
d’effet du tramadol au réveil qui est entre 20 min et 1 à 2 heures [6]. Les effets
du tramadol sur le tractus gastro-intestinal sont moins marqués que ceux de
la morphine à dose équi-analgésique [7]. Comme les autres bloqueurs de la
recapture de la sérotonine, le tramadol peut provoquer des convulsions aux
doses thérapeutiques. Ceci a été rapporté en cas d’antécédents d’épilepsie, de
prise concomitante d’antidépresseurs tricycliques ou d’autres substances qui
inhibent la recapture de la sérotonine, facteurs favorisants qui constituent des
contre-indications à la prise de tramadol.
Le profil pharmacologique du tramadol, notamment les interactions syner-
giques tramadol-paracétamol et tramadol-AINS [8], le rende particulièrement
intéressant en association avec ces substances non morphiniques pour le trai-
tement des douleurs modérément intenses, chez les patients hospitalisés mais
aussi en chirurgie ambulatoire. Chez le patient hospitalisé, le tramadol peut être
administré en PCA avec les réglages suivants : bolus de 20 mg, période interdite
de 10 min (5 à 15 min) et dose maximale sur 24 heures de 400 mg.
Il peut être aussi proposé pour soulager les douleurs postopératoires plus
intenses dans le cadre d’une analgésie multimodale. Dans ce cas, il est notam-
ment intéressant d’administrer le tramadol en perfusion continue et d’utiliser
une PCA de morphine en traitement de secours : administration peropératoire
de 1 mg.kg-1 de tramadol puis relais avec une perfusion de 0,2 mg.kg-1
.h-1 [9].

Douleur postopératoire 149
En conclusion, le tramadol est efficace pour traiter les douleurs postopéra-
toires modérées à sévères chez les patients hospitalisés ou ambulatoires. Son
mécanisme d’action est original. Il constitue un analgésique supplémentaire
intéressant qui peut être associé, dans le cadre d’une analgésie multimodale, à
d’autres analgésiques non morphiniques ou morphiniques. Ses effets indésira-
bles principaux sont les nausées et vomissements qui peuvent être prévenus en
débutant la dose de charge en peropératoire et en l’associant en postopératoire
à du dropéridol à dose faible.
2. CODÉINE
La codéine a une biodisponibilité de 60 %. Mais la codéine, en tant que
telle a une très faible affinité pour les récepteurs µ morphiniques, si bien que
l’action analgésique de la codéine est la conséquence de sa transformation en
morphine.
La codéine est métabolisée en morphine au niveau du cytochrome P450
et plus particulièrement du cytochrome 2D6, 10 molécules de codéine étant
transformées en une molécule de molécule de morphine par O-déméthylation.
Ainsi la codéine est 10 fois moins puissante que la morphine. Pour être efficaces
chez l’adulte, les formulations doivent contenir au moins 25 mg de codéine. Chez
l’enfant, la posologie maximale est de 6 mg.kg-1
.j-1 per os. Sept à 10 % de la
population métabolisent très lentement la codéine [10]. Chez de telles personnes
,
la codéine est inactive.
Comparé à un placebo en postopératoire, le NNT de l’association de 300 mg
de paracétamol et 30 mg de codéine est de 5,3, pour des doses doubles de
chacun des composants de l’association le NNT est de 3,1 [11]. Comparé au
paracétamol seul et pour la période postopératoire, le NNT de l’association
codéine-paracétamol varie entre 6,7 et 10 [11].
3. DEXTROPROPOXYPHÈNE
Le dextropropoxyphène est un morphinomimétique, agoniste des récep-
teurs morphiniques µ, considéré à tort comme responsable de moins d’effets
indésirables que la morphine et la codéine. Il ne s’agit en fait que d’un effet dû
aux très faibles doses administrées. Très peu d’études ont comparé l’association
dextropropoxyphène-paracétamol au paracétamol seul. En dépit de sa grande
utilisation, son bénéfice reste très controversé [12]. Ainsi, le NNT de l’association
65 mg de dextropropoxyphène-650 mg de paracétamol comparée à un placebo
est de 4,4, tandis que celui de 650 mg de paracétamol seul est de 5 et celui de
65 mg de dextropropoxyphène de 7,7 [13].
Par contre, le dextropropoxyphène induit les mêmes effets indésirables que
les autres morphinomimétiques notamment les nausées et les vomissements.
L’incidence de troubles neurologiques avec confusion a été particulièrement
rapportée chez le sujet âgé. Par ailleurs, en cas de surdosage, aux effets classi-
ques des morphiniques (apnées), le dextropropoxyphène a des effets toxiques
cardiaques (troubles du rythme par action sur la conductance sodique). Devant
un nombre élevé de cas de décès avant l’admission à l’hôpital par surdosage
au dextropropoxyphène en association avec de la prise d’alcool dans un but
d’autolyse [14], le dextropropoxyphène a été retiré récemment du marché au
Royaume Uni.

MAPAR 2006
150
4. NALBUPHINE (NUBAIN®)
La nalbuphine appartient à la classe des angonistes-antagonistes morphi-
niques. Elle exerce des propriétés agonistes des récepteurs K et antagonistes
des récepteurs µ.
La nalbuphine a un effet plafond sur l’analgésie. Il apparaît à partir d’une dose
entre 0,3 à 0,5 mg qui correspond à un équivalent entre 0,15 et 0,25 mg.kg-1 de
morphine. Cet effet plafond limite considérablement l’efficacité analgésique de
la nalbuphine sur les douleurs postopératoires intenses.
L’analgésie apparaît en 2 à 3 min après administration intraveineuse et en
15 à 20 min après injection intramusculaire ou sous-cutanée. Le pic de l’effet peut
être retardé jusqu’à 30 min après une administration intraveineuse. La durée de
l’analgésie est de 4 heures en moyenne.
L’action analgésique de la nalbuphine s’accompagne d’un certain degré de
dépression respiratoire. L’effet plafond de la nalbuphine sur la dépression respi-
ratoire apparaît à partir de 0,3 à 0,5 mg.kg-1, ce qui est identique à la dépression
respiratoire que crée 0,15 à 0,25 mg.kg-1 de morphine. Il correspond en moyenne
à une dépression de 50 % de la réponse ventilatoire au CO2. Contrairement à ce
qui a été noté avec la buprénorphine, la naloxone a la possibilité d’antagoniser
l’action dépressive respiratoire de la nalbuphine.
La nalbuphine est caractérisée par l’absence de modification hémodyna-
mique. Cette stabilité hémodynamique est particulièrement indiquée en cas
d’insuffisance cardiaque ou d’insuffisance coronarienne.
Comme les autres agonistes-antagonistes, la nalbuphine crée une sédation
plus marquée que les agonistes purs à dose équi-active. Les effets psychomo-
teurs, connus avec la pentazocine et le butorphanol s’observent également avec
la nalbuphine. La fréquence de ces effets indésirés est très supérieure à celle
rapportée avec les morphiniques agonistes purs.
Les nausées et les vomissements sont aussi fréquents qu’avec la morphine.
Ils peuvent apparaître de manière retardée, 2 h après l’injection. L’incidence est de
5 à 10 %. L’action de la nalbuphine sur les fibres musculaires lisses est moindre
que celle des morphinomimétiques. Le transit intestinal est peu modifié et la
pression dans les voies biliaires n’est pas augmentée de manière significative.
Cependant la nalbuphine crée le même retard à la vidange gastrique que la mor-
phine par augmentation du tonus du pylore. Néanmoins, l’absence d’effet marqué
sur le transit intestinal rend la nalbuphine très utile pour traiter les douleurs à
partir du deuxième jour postopératoire d’une chirurgie digestive.
La nalbuphine est peu toxicomanogène. Le syndrome de sevrage en cas
d’arrêt brutal d’une intoxication chronique est modéré, avec peu d’assuétude.
La nalbuphine est une substance à activité antagoniste quand elle est
administrée à la suite d’une substance agoniste, comme tous les morphiniques
de cette classe. Cette activité antagoniste est 25 fois moindre que celle de la
naloxone. Par contre, en cas d’inefficacité de la nalbuphine, la morphine peut
être utilisée immédiatement après.
La nalbuphine est présentée en ampoule de 20 mg sous forme de chlorhy-
drate. Les doses habituellement utilisées varient entre 0,20 et 0,40 mg.kg-1 toutes
les 4 à 6 heures. Au-delà de ces doses, l’activité analgésique n’augmente pas.

Douleur postopératoire 151
5. NÉFOPAM
Le néfopam est un analgésique central dont l’activité ne s’effectue pas par
l’intermédiaire des récepteurs opioïdes ou des prostaglandines. Le principal effet
analgésique du néfopam s’exerce au niveau de la recapture des monoamines.
Les tests in vitro sur des préparations de synaptosomes ont montré que le néfo-
pam inhibe la recapture de la sérotonine et de la noradrénaline. Récemment,
un effet du néfopam sur les canaux sodiques et calciques voltage-dépendants
présynaptiques a été mis en évidence modulant ainsi indirectement la libération
de glutamate, mais sans action directe sur les récepteurs NMDA [15, 16].
L’absence d’effet sur la fonction respiratoire ainsi que sur l’hémostase
procure un indéniable avantage au néfopam pour l’analgésie postopératoire. Le
néfopam ne provoque pas d’accoutumance, de dépendance ou de phénomène
de sevrage et son utilisation prolongée n’est pas suivie d’un épuisement de son
activité antalgique.
Le ratio d’équi-analgésie du néfopam avec la morphine est compris entre
1 : 2 et 1 : 3. Quinze à 20 mg de néfopam produisent une analgésie comparable
en puissance à celle produite par 50 mg de péthidine. Après chirurgie dentaire,
l’analgésie produite par 20 mg de néfopam est comparable à celle obtenue avec
75 mg de diclofenac [17].
En postopératoire de chirurgie orthopédique ou abdominale, l’administration
intramusculaire de néfopam permet de réduire de 25 à 50 % la consomma-
tion de morphine. Cette réduction est supérieure à celle du propacétamol. Le
néfopam est, comme pour les autres analgésiques, administré dans le cadre
d’une analgésie multimodale associant différents médicaments analgésiques.
L’association néfopam-AINS a été montrée particulièrement intéressante car
synergique [18].
Des effets indésirables sont signalés avec le néfopam et sont par ordre
décroissant de fréquence les sueurs, la somnolence, les manifestations
nauséeuses avec ou sans vomissements, les malaises, ainsi que des réactions
de type atropinique : sécheresse buccale, tachycardie, palpitations, vertiges,
rétention d’urines. La survenue de sueurs, notée lors de l’administration de
néfopam n’est pas la conséquence d’un effet thermorégulateur (la température
centrale à tendance à baisser lors de la prise de néfopam) mais plutôt d’une
stimulation directe des glandes eccrines. Ces différents effets indésirables sont
dépendants des concentrations plasmatiques de néfopam. Ils sont le fait d’ad-
ministrations trop rapides. Les administrations IV de 20 mg de néfopam doivent
être réalisées au moins sur 30 min. L’administration optimale est de 80 à 120 mg
en perfusion continue sur 24 heures sans dose de charge [19]. Si celle-ci devait
être réalisée, il faut privilégier une dose de charge de 20 mg en peropératoire
administrée durant au moins 30 min.
CONCLUSION
En conclusion, les médicaments analgésiques du palier 2 sont de mécanis-
mes d’action différents, mais ils sont très utiles associés à d’autres analgésiques
des niveaux 1 et 3 dans le contexte général d’une analgésie multimodale.
 6
6
1
/
6
100%