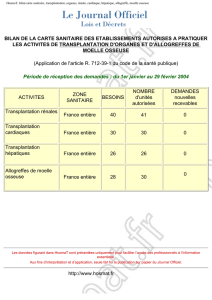
Lire l'article complet

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012
182
Dossier thématique
Récidive de la néphropathie
initiale après transplantation
rénale sur le greffon
Résumé
Summary
»La récidive de l’hyperoxalurie primitive de type 1 est constante
après transplantation rénale, avec un délai très variable, car elle ne
remplace que l’organe cible. Il est donc indispensable d’envisager
une transplantation combinée hépatique et rénale (simultanée
ou séquentielle) dans la plupart des cas, sauf peut-être lorsqu’il
existe une sensibilité documentée à la pyridoxine et une mutation
réputée sensible. En effet, seules l’hépatectomie du foie propre et
la greffe orthotopique du foie restaurent les fonctions de l’organe
source. La recherche actuelle s’oriente vers une alternative à la
greffe d’organes, encore illusoire.
Mots-clés : Hyperoxalurie primitive de type 1 – Récidive après trans-
plantation – Transplantation combinée hépatique et rénale.
Primary hyperoxaluria always leads to recurrence after
renal transplantation, within a variable interval of time.
Most patients should therefore receive a (simultaneous or
sequential) combined liver-kidney transplantation, except
maybe in selected subjects with evidence of pyridoxin
responsiveness and appropriate genotype. Indeed, native liver
hepatectomy followed by orthotopic liver transplantation is
the only option to provide adequate enzyme source. Current
research relies on alternatives to organ transplantation that
cannot yet be considered in humans.
Keywords: Primary hyperoxaluria type 1 – Post-transplant
recurrence – Combined liver-kidney transplantation.
Récidive de l’oxalose
après transplantation rénale
Post-transplant recurrence of primary hyperoxaluria type 1
Pierre Cochat*,**, Justine Bacchetta*, Anne-Laure Leclerc*
Le terme “oxalose” désigne l’atteinte systémique
presque constante dans l’évolution des hyper-
oxaluries primitives, et c’est dans cette situation
que la greffe d’organes trouve sa place (1). En France, la
plupart des patients traités présentent une hyperoxa-
lurie primitive de type 1 (HP1), mais, dans environ 10 %
des cas, il s’agit de types 2, 3 ou autres, indéterminés,
qui ne seront pas abordés ici, notamment du fait de
données trop anecdotiques en termes de transplan-
tation d’organes.
L’HP1 est une affection de transmission récessive auto-
somique qui concerne 1 naissance vivante sur 120 000
en Europe occidentale. Elle est la conséquence du
déficit d’une enzyme normalement produite au niveau
des peroxysomes hépatocytaires, l’alanine glyoxylate
amino transférase (AGT), dont la coenzyme est le phos-
phate de pyridoxine (vitamine B6). Les mutations du
gène AGXT sont nombreuses et généralement privées,
mais il existe plusieurs relations génotype-phénotype,
de sorte que la maladie s’exprime soit parce que la
production d’AGT par le peroxysome est déficiente
ou absente (déficit quantitatif), soit parce qu’elle est
délocalisée dans la mitochondrie, ce qui rend l’AGT
inefficace (déficit fonctionnel), les déficits quantitatifs
étant généralement plus graves que les déficits fonc-
tionnels (2). Ce déficit aboutit dans tous les cas à une
production massive d’oxalate, qui a d’abord des effets
sur le rein et les voies urinaires, puis sur l’ensemble
de l’organisme lorsque l’élimination de l’oxalate rénal
devient inférieure à sa production hépatique. Outre
les répercussions sur la qualité de vie, la mortalité
des patients atteints d’insuffisance rénale chronique
terminale (IRCT) est 3 fois plus élevée lorsque cette
dernière est due à une HP1 (3).
La récidive de la maladie sur le greffon rénal est fré-
quente et peut survenir dans 2 situations distinctes :
✓
en cas de greffe rénale isolée lorsque la source de
la production (foie) n’a pas été retirée ;
✓
lorsque l’accumulation systémique entraîne un
relargage d’oxalate avec dépôts dans le greffon rénal,
même en cas de greffe combinée hépatique et rénale (4).
Par ailleurs, la récidive sur un greffon rénal reste, de
nos jours, un mode de révélation de l’HP1 dans 10 %
des cas (2, 5, 6).
* Service de néphrologie,
rhumatologie et dermato-
logie pédiatriques,
hôpital Femme-Mère-
Enfant, Hospices civils
de Lyon, Bron.
** Centre de référence
des maladies rénales
rares Néphrogones,
IBCP-FRE 3310,
CNRS ; université Claude-
Bernard Lyon-I.

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012 183
Une fois l’HP1 confirmée par biologie moléculaire,
la stratégie de transplantation est dictée par le stade
de la maladie rénale chronique (MRC) et par l’éva-
luation du stock systémique d’oxalate qui s’ensuit,
estimé par la mesure répétée de la filtration glomé-
rulaire (FG), de l’oxalémie et de l’atteinte systémique
(fond d’œil, imagerie et histomorphométrie osseuse).
Cette atteinte systémique, grossièrement corrélée
au temps passé au-delà des stades 3 et 4 de la MRC,
conditionne la morbidité et la mortalité ; idéalement,
toute transplantation doit donc être une démarche
précoce, dès que la FG est inférieure à 40 à 30 ml/
mn/1,73 m2 (7).
Transplantation rénale :
une option à abandonner ?
La transplantation rénale permet une élimination effi-
cace de l’oxalate soluble ; cependant, le greffon doit
alors faire face non seulement à la production hépa-
tique d’oxalate soluble mais aussi à la part d’oxalate
tissulaire libérée dans le plasma. Il s’ensuit une récidive
des dépôts d’oxalate de calcium dans le greffon rénal
dans 100 % des cas et une pérennisation de la surcharge
tissulaire. Ainsi, la survie du greffon rénal en l’absence
de transplantation hépatique est de 46 % à 1 an, 28 %
à 3 ans et 14 % à 5 ans (3). La transplantation rénale
isolée est donc a priori contre-indiquée en raison du
risque de récidive rénale et de l’absence d’efficacité sur
l’atteinte systémique (8).
Chez certains patients porteurs de la mutation
Gly170Arg ou Phe152Ile, il existe un degré variable
mais certain de sensibilité à la pyridoxine ; celle-ci
doit donc être systématiquement testée et validée, et
peut alors conduire à envisager une transplantation
rénale isolée associée à un traitement ininterrompu
par pyridoxine (2). Toutefois, cette stratégie n’est pas
appliquée dans les centres experts et n’est concevable
qu’en cas de réponse totale à la pyridoxine (4).
Dans les pays en développement, la transplanta-
tion rénale isolée est proposée soit en l’absence de
diagnostic précis, soit dans l’attente d’une trans-
plantation combinée hépatique et rénale dans un
autre pays, soit, malheureusement, comme seule
alternative à la dialyse. Les échecs sont nombreux,
d’autant que les mutations associées à la sensibilité à
la pyridoxine sont peu fréquentes dans ces pays. Cela
pose d’importants problèmes éthiques et, parfois, de
santé publique (5). Ce constat permet d’insister sur la
nécessité d’entreprendre un traitement conservateur
(hydratation, citrate, pyridoxine) précoce et agressif,
mais aussi sur le fait qu’il est légitime de s’abstenir
de tout acharnement thérapeutique en l’absence de
ressources adaptées, notamment dans les formes
sévères d’oxalose infantile.
Argumentaire pour la transplantation
hépatique
Dans la mesure où la production d’oxalate est stricte-
ment limitée aux peroxysomes hépatocytaires, unique
site de détoxification du glyoxylate, seule l’hépatec-
tomie permet de contrôler la situation métabolique.
Cette production est si importante que les tentatives de
remplacement enzymatique sont actuellement toutes
vouées à l’échec tant que le foie natif est présent ; cer-
tains espoirs sont néanmoins permis avec la greffe
d’hépatocytes (9, 10).
La transplantation hépatique orthotopique représente
donc une forme d’enzymothérapie substitutive en
apportant l’enzyme manquante au bon endroit, tant en
ce qui concerne l’organe (foie) que la cellule (hépato-
cyte) ou la structure intracellulaire (peroxysome).
L’objectif de la transplantation d’organes dans l’HP1
est donc de transformer un bilan d’oxalate inexora-
blement positif en un bilan progressivement négatif,
en interrompant la production hépatique (transplan-
tation hépatique) et en restaurant l’épuration rénale
(transplantation rénale).
De manière anecdotique, l’utilisation du foie d’un “rece-
veur-donneur” atteint d’HP1 dans le cadre d’une greffe
domino s’est soldée par une insuffisance rénale rapide
du fait de la précipitation massive d’oxalate dans le rein
propre du receveur non atteint d’HP1 (11).
Transplantation combinée hépatique
etrénale
L’HP1 représente actuellement la principale indi-
cation de transplantation combinée hépatique
et rénale (12, 13). Les résultats sont globalement
encourageants, tant chez l’enfant que chez l’adulte,
avec une survie des patients de 86 % à 1 an, 79 % à
3 ans et 76 % à 5 ans ; quant à la survie rénale, elle
est respectivement de 82, 79 et 76 % (3). Toutefois, la
transplantation combinée chez des patients en IRCT
avec thésaurismose significative expose au risque de
compromettre la fonction et la survie du greffon rénal
soumis à une élimination massive et brutale d’oxalate
dès le déclampage vasculaire (oxalate soluble circu-
lant + oxalate libéré des tissus). Cette stratégie doit
donc être considérée et préparée relativement tôt
dans l’évolution de l’insuffisance rénale, afin de limiter
justement un stockage excessif (8). La transplantation
Récidive de l’oxalose

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012
184
Dossier thématique
Récidive de la néphropathie
initiale après transplantation
rénale sur le greffon
combinée peut être réalisée en un seul temps (foie et
rein) ou de manière séquentielle (foie puis rein ; mais
pas l’inverse).
Transplantation préemptive isolée du foie
Théoriquement, il est légitime de proposer une trans-
plantation hépatique isolée lorsque la FG est encore
suffisamment bonne pour laisser espérer une stabili-
sation ou une amélioration après une greffe (14, 15).
Cette stratégie a été appliquée ponctuellement à des
receveurs dès le stade 3 de la MRC, avec des résultats
variables, et en tout cas suffisamment aléatoires pour
en limiter les indications. En effet, l’évolution est dif-
férente pour chaque malade, et, une fois atteint le
stade 3 de la MRC, le délai avant le stade terminal est
très variable d’un sujet à l’autre, de sorte que la trans-
plantation hépatique peut introduire un risque vital
prématuré. Ce risque n’est pas comparable au risque
fonctionnel lié à la progression de la thésaurismose mais
impose d’éviter ou de limiter la surcharge systémique.
Les indications sont donc très limitées et doivent être
discutées par des experts (4).
En outre, il est important de rappeler que l’HP1 est
la seule maladie péroxysomale qui ne comporte pas
d’atteinte neurologique, et pour laquelle le traitement
conservateur s’est considérablement amélioré et a été
bien codifié au cours des 10 dernières années ; la notion
d’“urgence métabolique” n’intervient donc pas dans
ce cadre.
Évolution systémique
aprèstransplantation
Après transplantation combinée hépatique et rénale,
l’oxalémie se normalise d’autant plus lentement que
le stockage tissulaire était important, nécessitant
des mois, voire des années. Le risque de néphrocal-
cinose et de lithiase est donc transposé au greffon
rénal. Aussi, lorsque l’exposition présumée du greffon
rénal est trop importante (oxalémie > 50 µmol/l), des
séances quotidiennes d’hémodialyse sont encore
nécessaires, à condition que les variations volé-
miques soient contrôlées pour ne pas provoquer,
après la transplantation, une oligurie qui pourrait
augmenter brutalement la saturation urinaire en
oxalate de calcium, et donc le risque de cristallisa-
tion. De même, l’hémodialyse après la transplantation
s’impose en cas de retard de fonction du greffon afin
de ne pas cumuler nécrose tubulaire aiguë et pré-
cipitation parenchymateuse d’oxalate de calcium.
En revanche, lorsque la diurèse est immédiate et
suffisamment abondante après la transplantation,
l’hémodialyse est inutile, et il est alors essentiel de
protéger le greffon rénal par une hydratation abon-
dante (2 à 3 l/1,73 m
2
/j), des inhibiteurs de la cris-
tallisation (citrate, 100 à 150 mg/kg/j) et, parfois, un
diurétique thiazidique en cas d’hypercalciurie (hydro-
chlorothiazide, 1 à 2 mg/kg/j) [8]. En revanche, il n’y a
pas lieu de poursuivre le traitement par pyridoxine,
dans la mesure où le foie a été greffé. Le suivi bio-
logique repose alors essentiellement sur l’évolution
de l’oxalémie et du volume cristallin.
Après une transplantation combinée réussie, l’amélio-
ration systémique peut être spectaculaire : amélioration
de l’atteinte ostéoarticulaire, de la fonction myocar-
dique, des manifestations artéritiques et, surtout, de
la qualité de vie globale.
Quelle stratégie de transplantation
proposer en 2012 ?
Plusieurs cas de figure peuvent être envisagés (8, 14).
✓
Chez les patients dont la FG est supérieure à 30 à
40 ml/mn pour 1,73 m2 et pour lesquels une indication
de transplantation hépatique isolée pourrait être discu-
tée, il semble préférable d’attendre que la FG diminue
davantage afin de proposer une transplantation com-
binée hépatique et rénale en un temps.
✓Chez les patients suivis régulièrement et dont la
FG diminue progressivement, une transplantation
combinée hépatique et rénale en un temps doit
être considérée au stade 3b de la MRC et réalisée
idéalement au stade 4, avant le stade terminal, pour
éviter une transplantation à un stade trop avancé.
Il s’agit alors généralement d’un donneur décédé
unique, ce qui constitue un avantage immunologique
et logistique.
✓
Chez les patients ayant atteint le stade d’IRCT et
traités par dialyse, l’épuration doit tout d’abord être
optimisée (hémodialyse quotidienne ± dialyse périto-
néale) avant d’envisager une transplantation combinée
hépatique et rénale. Toutefois, à ce stade, la transplan-
tation peut être envisagée en 3 temps :
• transplantation hépatique ;
•
période d’épuration optimale afin de stabiliser l’oxa-
lémie en dessous de 50 µmol/l ;
• transplantation rénale.
Cette séquence pose problème sur le plan logistique,
mais est rationnelle du point de vue biochimique.
Cela concerne notamment les jeunes enfants qui pré-
sentent une forme infantile sévère. Le recours à un
donneur vivant n’est pas exclu dans ce contexte de
transplantation en 2 temps, soit pour un seul organe,

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012 185
soit pour les 2 organes (16) ; le donneur vivant apparenté
peut être hétérozygote pour PH1 et paucisymptoma-
tique, ce qui nécessite une évaluation minutieuse et
spécifique avant la greffe. Si la transplantation com-
binée hépatique et rénale est réalisée en un seul temps,
l’épuration de l’oxalate doit être impérativement assurée
efficacement par le greffon ou, à défaut, par une hémo-
dialyse périopératoire.
Le choix de la stratégie dépend évidemment des
moyens disponibles et de l’expérience de l’équipe,
mais tout doit être fait pour que le temps passé
au-delà du stade 3 de la MRC soit le plus bref possible.
La lourdeur de la prise en charge impose un accom-
pagnement du patient dès le moment du diagnostic,
afin de lui permettre de comprendre au mieux sa
maladie pour qu’il puisse en saisir les risques et les
enjeux thérapeutiques.
Conclusion
Le problème de la récidive de la maladie initiale après
une transplantation d’organes pour oxalose ne devrait
plus concerner la transplantation rénale isolée, car les
indications en sont devenues confidentielles. Toutefois,
l’absence de diagnostic précis avant une transplan-
tation rénale de novo expose encore au risque de ne
diagnostiquer une HP1 qu’après récidive et perte du
greffon. Dès lors que la transplantation hépatique
est envisagée, toutes les mesures diagnostiques et
thérapeutiques doivent être prises pour éviter – ou,
au moins, limiter – les dépôts rénaux d’oxalate issus du
relargage systémique, ce qui pose le problème d’une
priorité spécifique sur liste d’attente. Le recours à des
thérapeutiques substitutives permettant d’eviter ou de
différer la greffe d’organes est séduisant mais relève
encore du domaine de la recherche. ■
1. Beck BB, Habbig S, Dittrich K et al. Liver cell transplantation
in severe infantile oxalosis – a potential bridging procedure
to orthotopic liver transplantation? Nephrol Dial Transplant
2012;27(7):2984-9.
2. Cochat P, Fargue S, Bacchetta J, Bertholet-Thomas A,
Sabot JF, Harambat J. Hyperoxalurie primitive. Nephrol Ther
2011;7:249-59.
3. Cochat P, Fargue S, Harambat J. Primary hyperoxaluria
type 1: Strategy for organ transplantation. Curr Opin Organ
Transplant 2010;15:590-3.
4. Cochat P, Hulton SA, Acquaviva C et al. Primary hyperoxalu-
ria Type 1: indications for screening and guidance for diagnosis
and treatment. Nephrol Dial Transplant 2012;27:1729-36.
5. Cochat P, Liutkus A. Preemptive liver transplantation for pri-
mary hyperoxaluria type 1. Acta Gastroenterol Belg 2005;68:74-5.
6. Darwish AA, McKiernan P, Chardot C. Paediatric liver
transplantation for metabolic disorders. Part 1: Liver-based
metabolic disorders without liver lesions. Clin Res Hepatol
Gastroenterol 2011;35:194-203.
7. Galanti M, Contreras A. Excellent renal function and reversal
of nephrocalcinosis 8 years after isolated liver transplantation
in an infant with primary hyperoxaluria type 1. Pediatr Nephrol
2010;25:2359-62.
8. Harambat J, Fargue S, Acquaviva C et al. Genotype-
phenotype correlation in primary hyperoxaluria type 1: the
p.Gly170Arg mutation is associated with a better outcome.
Kidney Int 2010;77:443-9.
9.
Harambat J, van Stralen KJ, Espinosa L et al. Characteristics
and outcomes of children with primary oxalosis requiring renal
replacement therapy. Clin J Am Soc Nephrol 2012;7:458-65.
10.
Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias.
Kidney Int 2009;75:1264-71.
11.
Jiang J, Salido E, Guha C et al. Correction of hyperoxaluria
by liver repopulation with hepatocytes in a mouse model of
primary hyperoxaluria type 1. Transplantation 2008;85:1253-60.
12. Madiwale C, Murlidharan P, Hase NK. Recurrence of primary
hyperoxaluria: an avoidable catastrophe following kidney
transplant. J Postgrad Med 2008;54:206-8.
13. Malla I, Lysy PA, Godefroid N et al. Two-step transplan-
tation for primary hyperoxaluria : cadaveric liver followed by
living donor related kidney transplantation. Pediatr Transplant
2008;13:782-4.
14. Mehrabi A, Fonouni H, Ayoub E et al. A single center expe-
rience of combined liver kidney transplantation. Clin Transplant
2009;23(Suppl. 2):102-14.
15. Saner FH, Treckmann J, Pratschke J et al. Early renal fai-
lure after domino liver transplantation using organs from
donors with primary hyperoxaluria type 1. Transplantation
2010;90:782-5.
16. Spasovski G, Beck BB, Blau N, Hoppe B, Tasic V. Late diagno-
sis of primary hyperoxaluria after failed kidney transplantation.
Int Urol Nephrol 2010;42:825-9.
Références bibliographiques
Récidive de l’oxalose
1
/
4
100%