2017TOU33028

2
UNIVERSITE TOULOUSE III – PAUL SABATIER
FACULTE DE CHIRURGIE DENTAIRE
Année 2017 Thèse n°2017-TOU3-3028
THESE
POUR LE DIPLOME D’ETAT DE DOCTEUR EN CHIRURGIE
DENTAIRE
Présentée et soutenue publiquement par
PONS GERMAIN Marie-Sophie
Le 13 Mars 2017
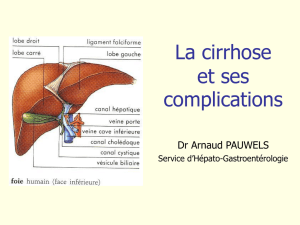
RELATION BIDIRECTIONNELLE ENTRE FONCTIONS
HEPATIQUES ET CAVITE BUCCALE
Directeur de thèse : Docteur BLASCO-BAQUE Vincent
JURY
Président Professeur DIEMER Franck
Assesseur Docteur GURGEL-GEORGELIN Marie
Assesseur Docteur LAURENCIN-DALICIEUX Sara
Assesseur Docteur BLASCO-BAQUE Vincent

3

4

5
A ma famille, mes amis…

6
A notre président du jury,
Monsieur le Professeur DIEMER Franck :
- Professeur des Universités, Praticien Hospitalier d’Odontologie
- Responsable de la sous-section d’Odontologie Conservatrice, Endodontie
- Docteur en Chirurgie Dentaire,
- D.E.A. de Pédagogie (Education, Formation et Insertion) Toulouse Le Mirail,
- Docteur de l’Université Paul Sabatier,
- Responsable du Diplôme Inter Universitaire d’Endodontie à Toulouse,
- Habilitation à diriger des recherches (H.D.R.),
- Vice- Président de la Société Française d’Endodontie
- Lauréat de l'Université Paul Sabatier
Nous vous sommes très reconnaissants d’avoir accepté la
présidence de notre jury de thèse. Nous vous remercions pour votre
enseignement théorique et pratique ainsi que votre disponibilité
durant l’ensemble de nos études. Veuillez trouvez ici notre plus
grand respect et l’expression de notre gratitude.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
1
/
86
100%