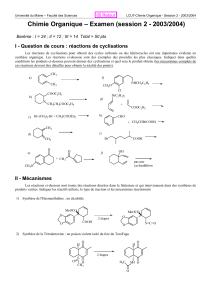
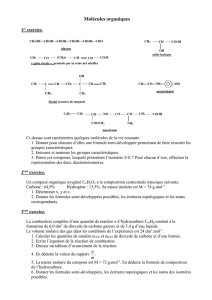
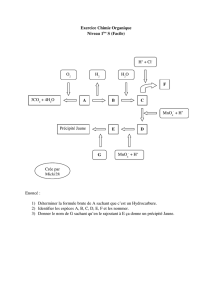
Modèles d`enzymes

1
Modèles d’enzymes
• systèmes conçus pour mimer les substrats et les catalyseurs des réactions enzymatiques
• facilitent l’étude mécanistique d’une réaction similaire à la réaction enzymatique
• informations mécanistiques ainsi accueillies peuvent avancer notre compréhension de la
catalyse enzymatique et nous permettre de concevoir d’autres catalyseurs
• section organisée selon les enzymes étudiées, leurs mécanismes natifs et leurs modèles
d’enzyme représentatifs
Références
A. Fersht, Enzyme Structure and Mechanism
R.B. Silverman, The Organic Chemistry of Enzyme-Catalyzed Reactions
Voet & Voet, Biochemistry

2
Phosphodiesterase
• catalyse l’hydrolyse des liens P-OR dans des phosphodiesters : (RO)P(O)O-
• enzyme digestive qui catalyse l’hydrolyse de l’ARN en nucléotides
Ribonucléase A
• première enzyme dont la séquence a été déterminée en 1960 (seulement 124 acides aminés)
• synthétisée par Merrifield en 1969
• Moore et Stein ont gagné le Prix Nobel en 1972 pour les techniques de séquençage qu’ils ont
développées pour déterminer la séquence de la RNase A
• l’ARN est clivé avec préférence (spécificité) pour une base de côté 3' de pyrimidine (uracile
ou cytosine)
Site actif de la ribonucléase A.
Notez l’orientation des groupes imidazoles des résidus His12 et His119.
Mécanisme
• l’ARN est clivé, mais pas l’ADN, à cause de la cyclisation avec 2'-OH qui a lieu, suivie par
l’hydrolyse lente du phosphate cyclique intermédiaire
• profil pH-vitesse : deux valeurs de pKa provenants du kcat / KM vs pH : 5.22 et 6.78 (enzyme
libre) ou 6.3 et 8.1 provenants du kcat vs pH (complexe enzyme-substrat)
• deux mécanismes ont été proposés pour la réaction catalytique : un mécanisme
« concomitant » et un mécanisme « séquentiel »

3
• mécanisme « concomitant » :
O
O
POO
OR'
O
R
O
POO
O
CH2
O
POO
O
CH2
OH
C
A
OH
O
R
O
POO
O
CH2
OO
C
P
OO
NH
N
His12 NH
NH His12
His119
NH NH
NH N
His119
HOH
O
R
O
POO
O
CH2
O
POO
O
OH
C

4
• mécanisme « séquentiel » :
O
O
POO
OR'
O
R
O
POO
O
CH2
O
POO
O
CH2
OH
C
A
OH
NH
N
His12
NH N
His119
O
O
POO
OR'
O
R
O
POO
O
CH2
O
POHO
O
CH2
OH
C
A
OH
O
R
O
POO
O
CH2
OO
C
O
O
POO
OR'
P
O
CH2A
OH
OOH
NH
NH
His12
His119
NH NH
O
R
O
POO
O
CH2
OO
C
O
O
POO
OR'
P
O
CH2A
OH
OOH
NH NH
His119
triester intermédiaire
O
R
O
POO
O
CH2
OO
C
P
OOH

5
Modèle de Breslow
• site de liaison fonctionnalisé avec des groupements catalytiques
• figure la liaison d’un modèle de substrat par un modèle d’enzyme (« enzyme artificielle ») (J.
Mol. Cat. 1994, 91, 161-174)
Cyclodextrine
• polymère cyclique du α-D-glucopyranose
• la cyclodextrine peut comprendre six (α), sept (β) ou huit (γ) unités de glucose
• son intérieure est relativement hydrophobe, son extérieure est relativement hydrophile
(soluble dans l’eau)
• forme un cavité avec des groupements OH primaires autour du bout étroit et des groupements
OH secondaires situés au bout plus large
• diamètre interne du β-CD est ~7.0 Å (parfait pour l’inclusion d’un anneau aromatique)
• groupes fonctionnels (OH) rendent le CD plus susceptible à modification synthétique, pour
introduire des groupes réactifs
O
OH
HO O
OH
OOH
HO
O
OH
O
OH
OH
O
HO
O
HO
OH
O
HO
O
HO OH
O
HO
O
HO
OH
O
OH
O
OH
HO
O
OH
β-cyclodextrine :
(1,4-α-D-glucose)7
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
1
/
43
100%