Chapitre 16

Faculté de Médecine de Strasbourg, Module de Pharmacologie Clinique DCEM3 2011/2012
« Les antipaludéens » Dr D Filisetti & Pr L Monassier Mise à jour janvier 2012
1/15
Chapitre 16 : LES ANTIPALUDEENS
Item 99 : Paludisme, argumenter l’attitude thérapeutique et planifier le suivi du patient
Plan :
Introduction
A. Classification des antipaludéens (= anti-malariques)
B. Schizonticides érythrocytaires
1) Quinolones et dérivés :
1.1. Chloroquine (Nivaquine®)
1.2. Quinine
1.3. Méfloquine (Lariam®)
2) Artémisine et dérivés
C. Schizonticides érythrocytaires et tissulaires
1) Antifoliques, antifoliniques et atovaquone
2) Doxycycline (Doxypalu®)

Faculté de Médecine de Strasbourg, Module de Pharmacologie Clinique DCEM3 2011/2012
« Les antipaludéens » Dr D Filisetti & Pr L Monassier Mise à jour janvier 2012
2/15
INTRODUCTION
Le paludisme (ou malaria en anglais) est une des maladies infectieuses les plus fréquentes dans le
monde. En 2008, 40 % de la population mondiale vit dans des zones impaludées (voir les sites internet
http://www.rbm.who.int/fr/index.php et http://www.who.int/topics/malaria/fr/). Le paludisme a touché 216
millions de personnes et causé le décès de 655 000 d’entre elles en 2010.
Le paludisme est dû à une infection par certains parasites du genre Plasmodium. Les espèces
concernées sont falciparum, vivax, ovale, malariae et knowlesi. Les infections à P. vivax, ovale et
malariae sont habituellement mineures, les formes graves de paludisme sont dues à Plasmodium
falciparum.
Plasmodium est transmis par un moustique du genre Anopheles, seules les anophèles femelles sont
hématophages.
Le cycle évolutif de Plasmodium comporte une phase sexuée ou sporogonique qui se déroule chez le
moustique et une phase asexuée ou schizogonique qui se déroule chez l’Homme. Cette phase
schizogonique a un stade pré-érythrocytaire (hépatique) et un stade érythrocytaire. Plasmodium se
multiplie dans le foie et libère des formes appelées mérozoïtes qui pénètrent dans les globules rouges. Le
parasite utilise l’hémoglobine pour son développement, qu’il ingère dans sa vacuole.
Plasmodium falciparum est devenu résistant à de nombreux antipaludéens (ex. chloroquine, et à d’autres
antipaludéens selon son origine géographique : méfloquine, sulfadoxine, pyriméthamine, …).
Ce cours envisage les bases pharmacologiques des médicaments antipaludéens pour la prise en charge
des accès palustres et la chimioprophylaxie du paludisme.
A. CLASSIFICATION DES ANTIPALUDEENS (OU ANTI-MALARIQUES)
Selon le mode d’action et la structure chimique, la classification suivante est classique :
Schizonticides érythrocytaires :
• Amino-4-quinoléine : chloroquine, amodiaquine
• Arylamino-alcools :
o quinoléine méthanols : quinine, méfloquine
o quinghaosu et dérivés de l’artémisinine (artéméther, artésunate, dihydroartémisinine)
Les antipaludéens à effet intra-érythrocytaire traversent la membrane des globules rouges puis celle du
parasite et pénètrent dans sa vacuole digestive où ils s’accumulent. La résistance aux antipaludéens
intra-érythrocytaires serait due à des expulsions des antipaludéens par les parasites.
Schizonticides érythrocytaires et tissulaires :
• Antifoliques : sulfamides (sulfadoxine)
• Antifoliniques : pyriméthamine, proguanil
• Antibiotiques : doxycycline

Faculté de Médecine de Strasbourg, Module de Pharmacologie Clinique DCEM3 2011/2012
« Les antipaludéens » Dr D Filisetti & Pr L Monassier Mise à jour janvier 2012
3/15
B. SCHIZONTICIDES ERYTHROCYTAIRES
1. Quinolone et dérivés
1.1. Chloroquine (Nivaquine®)
1.1.1 Mécanisme d’action
La chloroquine traverse la membrane de l’érythrocyte parasité et se concentre fortement dans la vacuole
digestive du parasite. La chloroquine entraîne ainsi une accumulation, toxique pour le Plasmodium, de
molécules d’hématine, produit de dégradation de l’hème de l’hémoglobine, qui entraîne la lyse du
parasite.
Résistance : liée à une incapacité à accumuler la chloroquine dans la vacuole digestive du parasite,
phénomène lié à un efflux de chloroquine de la vacuole (mutation du gène PfCRT).
1.1.2. Pharmacocinétique
Biodisponibilité orale : satisfaisante mais variable, améliorée par la prise concomitante d’aliments.
Diffusion : facile, fixation dans les érythrocytes et les tissus riches en mélanine (choroïdes, corps ciliaires)
d’où une demi-vie d’élimination longue de 20 à 60 jours. 60 % de liaison aux protéines plasmatiques.
Métabolisme : par CYP 3A4, 2D6.
Elimination : rénale d’où baisse de posologie chez les insuffisants rénaux.
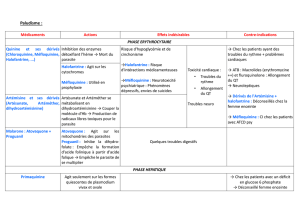
1.1.3. Indications :
• Traitement des accès palustres à Plasmodium vivax, ovale et malariae : 600 mg en 1 prise puis
300 mg 6 h après et 300 mg le 2ème et 3ème jour, c’est également le traitement des accès simples
à P. falciparum en zone 1.
• Prophylaxie en zone 1 chloroquino-sensible : le jour du départ, pendant le séjour et 4 semaines
après le retour.
1.1.4. Contre-indications :
• Rétinopathie,
• Attention chez les patients atteints de porphyrie intermittente1, la chloroquine peut provoquer une
crise aiguë,
• Pour les patients ayant un déficit en G6PD2 (glucose 6 phosphate déshydrogénase) : possibilité
que la chloroquine déclenche une anémie hémolytique mais rare à doses thérapeutiques.
1.1.5. Effets indésirables :
La chloroquine peut provoquer une crise chez les patients atteints de porphyrie intermittente1 et une
anémie hémolytique chez les patients ayant un déficit en G6PD2 (glucose 6 phosphate déshydrogénase).
Effets indésirables rares dans le cadre d’une chimio-prophylaxie (moins d’ 1 effet grave sur 100 000
traitements). Les signes d’une toxicité grave surviennent en cas de surdosage, d’intoxication, ou de
traitements très prolongés (par ex. en rhumatologie).
Effets indésirables en cas de surdosage : troubles ophtalmologiques (troubles de l’accommodation,
rétinopathies, opacités cornéennes, dégénérescence maculaire), troubles auditifs (acouphène, surdité),
cardiovasculaires (allongement de l’espace QT, troubles de la conduction, choc cardiogénique), cutanés
(prurit, photosensibilisation, éruptions), hématologiques (effet myélo-suppresseur dose-dépendant) ou
neurologiques (exceptionnellement troubles du comportement ou convulsions).
1 Liste des médicaments susceptibles de provoquer une crise en cas de porphyrie intermittente : www.drugs-poprhyria.org
2 Déficit en G 6PD : www.gs-im3.fr

Faculté de Médecine de Strasbourg, Module de Pharmacologie Clinique DCEM3 2011/2012
« Les antipaludéens » Dr D Filisetti & Pr L Monassier Mise à jour janvier 2012
4/15
1.1.6. Précautions :
Chez les insuffisants rénaux, insuffisants hépatiques, patients déficitaires en G6PD, patients atteints de
porphyrie intermittente, patients ayant une myasthénie grave, épilepsie.
1.1.7. Interactions médicamenteuses :
Précautions d’emploi :
• + Topiques gastro-intestinaux anti-acides : diminution de l’absorption de chloroquine.
• + Ciclosporine : augmentation des concentrations de ciclosporine.
1.2. Quinine
Alcaloïde extrait de l’écorce d’un arbre tropical le quinquina. Elle est présentée sous forme de plusieurs
spécialités pour administration injectable ou orale (voir liste ci-dessous). Pour le choix de posologie, il est
important de connaître les concentrations en quinine-base.
Attention, certaines spécialités contiennent des sulfites comme excipients pouvant entraîner ou aggraver
des réactions anaphylactiques.
Spécialité
Produit actif
Présentation-composition
Equivalence en
quinine-base et
proportion
Quinoforme®
Formiate de quinine
Solution injectable : ampoules
de 2 ml contenant 500 mg de
sel
219 mg/ml soit 87,6 %
Quinimax solution
injectable®
Gluconate de quinine
et de quinidine,
chlorhydrate de
cinchonine et de
cinchonidine
Solution injectable : ampoules
de 1 ; 2 ; 4 ml contenant par
ml : gluconate de quinine :
192,56 mg ; gluconate de
quinidine : 5,29 mg ;
chlorhydrate de cinchonine :
1,06 mg et chlorhydrate de
cinchonidine : 1 mg
120 mg/ml soit 62,3 %
d’alcaloïdes-base
Quinimax comprimés®
Chlorhydrate de
quinine et de quinidine,
de cinchonine et de
cinchonidine
Comprimés : chlorhydrate de
quinine : 587,25 mg ;
chlorhydrate de quinidine :
15,42 mg ; chlorhydrate de
cinchonine : 4,24 mg et
chlorhydrate de cinchonidine :
4,03 mg
480 mg/comprimé soit
81,7 % (500 mg
d’alcaloïdes-base par
comprimé)
Dichlorhydrate de
quinine®
Dichlorhydrate de
quinine
Solution injectable :
ampoules de 10 ml contenant
100 mg ou 300 mg de sel
Solution à 1 % : 8,17 %
mg/ml
Solution à 3 % : 24,5
mg/ml
Quinine®
Chlorhydrate de
quinine
Comprimés à 250 mg et 500
mg de sel
204 mg/comprimé et
409 mg/comprimé soit
81,7 %
1.2.2. Mécanisme d’action
La quinine, tout comme la chloroquine, se concentre dans la vacuole digestive de Plasmodium mais à
des concentrations moindres que la chloroquine. Comme pour la méfloquine, une fixation sur des sites
protéiques du parasite pourrait être favorisée par la lipophile du médicament.
1.2.3. Pharmacocinétique

Faculté de Médecine de Strasbourg, Module de Pharmacologie Clinique DCEM3 2011/2012
« Les antipaludéens » Dr D Filisetti & Pr L Monassier Mise à jour janvier 2012
5/15
Absorption : rapide par voie orale ou par voie intra-musculaire, biodisponibilité : 80 %, T1/2
≈
11 h.
Métabolisme : par CYP 3A4.
Elimination : rénale : 20 % sous forme inchangée, 80 % sous forme de métabolites dont un actif, d’où
adaptation de posologie chez les insuffisants rénaux.
La pharmacocinétique de la quinine est modifiée significativement par l’accès palustre : parfois
augmentation des concentrations de quinine à la suite de diminution du volume apparent de distribution et
de la clairance hépatique et rénale de la quinine.
1.2.4. Indications
Traitement de l’accès grave de paludisme à Plasmodium falciparum (par voie IV puis par voie orale
pendant 7 jours), des accès simples de paludisme à P. falciparum en zone 2 et 3 (2ème ligne), de tout
accès de paludisme (quelque soit l’espèce en cause) en cas de vomissements, traitement utilisable chez
la femme enceinte ou qui allaite. Non utilisé en prophylaxie.
1.2.5. Effets indésirables
Ils sont dose-dépendants :
• Fréquent, peu grave : cinchonisme (= acouphènes, vertiges, céphalées, troubles de la vision, baisse
aigüe de l’acuité auditive, troubles digestifs, vasodilatation périphérique).
• Hypoglycémie par augmentation de la sécrétion d’insuline.
Les signes de toxicité surviennent pour des concentrations plasmatiques élevées lors d’accès palustres
graves. Ce sont principalement :
• Toxicité cardiovasculaire : trouble de la conduction, troubles du rythme, allongement de l’espace
QT,
• Toxicité oculaire perte passagère de vision par atteinte des cellules rétiniennes,
• Toxicité auditive : altération de l’audition pour des fréquences élevées, acouphènes,
• Toxicité neurologique : vertiges,
• Toxicité cutanées photosensibilisation,
• Toxicité hématologique.
1.2.6. Précautions
En cas de déficit en G6PD2 : anémie hémolytique.
1.2.7. Interactions médicamenteuses
Peu documentées et ne semblent pas représenter de problème préoccupant pour traiter les accès
palustres.
Contre-indications : + Astémizole : risque majoré de troubles du rythme ventriculaire, notamment de
torsades de pointes (diminution du métabolisme hépatique de l'antihistaminique par la quinine).
1.3. Méfloquine (Lariam®)
1.3.1. Mécanisme d’action
Commun avec celui de la chloroquine, a une activité schizonticide sur Plasmodium falciparum
chloroquino-résistant (mais pas en zone 3+ car résistance par mutations dont l’hypothèse est qu’elles
conduiraient à l’expulsion du médicament de la vacuole du parasite).
1.3.2. Pharmacocinétique
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%