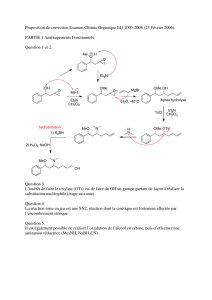
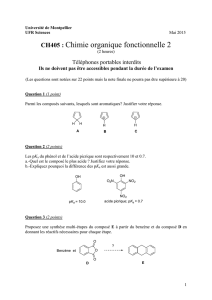
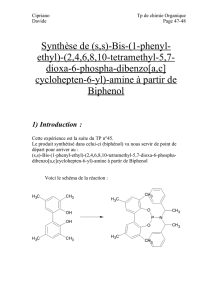
Désaromatisation hydroxylante de phénols par des réactifs iodés

Université Bordeaux 1
Les Sciences et les Technologies au service de l’Homme et de l’Environnement
N° d’ordre : 3924
THÈSE
PRÉSENTÉE A
L’UNIVERSITÉ BORDEAUX 1
ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES
Par Mélanie MARGUERIT
POUR OBTENIR LE GRADE DE
DOCTEUR
SPÉCIALITÉ : Chimie Organique
Désaromatisation Hydroxylante de Phénols par des Réactifs Iodés
Hypervalents : Application à la Synthèse de Substances Naturelles
Directeur de recherche : Pr. Stéphane QUIDEAU
Soutenue le : 11 décembre 2009
Après avis de :
M. WONG, Y. -S. Chargé de Recherche au CNRS, HDR, Université Grenoble Rapporteur
M. COSSIO, F. P. Professeur, Université du Pays Basque, Saint-Sébastien Rapporteur
Devant la commission d’examen formée de :
M. WONG, Y. -S. Chargé de Recherche au CNRS, HDR, Université Grenoble Rapporteur
M. COSSIO, F. P. Professeur, Université du Pays Basque, Saint-Sébastien Rapporteur
M. EATHERTON, A. J. GlaxoSmithKline, Harlow, Angleterre Examinateur
M. POUYSEGU, L. Maître de Conférences, Université Bordeaux 1 Examinateur
M. GUICHARD, G. Directeur de Recherche au CNRS, Université Bordeaux 1 Président de jury
M. QUIDEAU, S. Professeur, Université Bordeaux 1 Directeur de thès

2

Remerciements
3
Remerciements
Je tiens à remercier tout d’abord le Professeur Stéphane Quideau pour m’avoir accueillie au
sein de son équipe et pour m’avoir donné les moyens, comme il le dit lui-même, de « casser
les murs » qui se dressaient face à moi pour remplir les challenges qui m’étaient fixés.
Je souhaite également témoigner toute ma reconnaissance au Docteur Laurent Pouységu, qui
m’a encadrée pendant ces trois années, pour ses conseils scientifiques mais aussi pour le
modèle d’organisation qu’il est et qu’il a su m’insuffler, sans oublier son coaching et le temps
qu’il a consacré à la préparation de diverses présentations et à la relecture de mon manuscrit
de thèse.
Je remercie aussi Andrew Eatherton, qui a été mon co-directeur de thèse et qui a su apporter
un regard, à la fois critique et encourageant, sur les différents projets notamment au cours des
trois mois passés à ses cotés à GlaxoSmithKline. Ses précieux conseils ont été d’une grande
aide et sa bonne humeur chantante au laboratoire aura été une grande source d’énergie et
d’enthousiasme, avant de commencer ma dernière année de thèse.
Je tiens à exprimer mes sincères remerciements au Docteur Yung-Sing Wong et au Professeur
Fernando Cossio d’avoir accepté de juger ce travail en qualité de rapporteurs, ainsi qu’au
Professeur Gilles Guichard pour avoir accepté de présider le jury.
J’exprime ma gratitude aux chimistes de RMN et de spectrométrie de masse de l’IECB et de
GlaxoSmithKline pour leurs conseils pertinents et leur grande disponibilité tout au long de ma
thèse et en particulier Katell Bathany qui a été d’une aide précieuse notamment pour la
finalisation du projet des iodes chiraux. Je souhaite également remercier Patrick Pardon et
Rémi Jacquet pour leurs conseils avisés et leur aide en CLHP.
Un grand merci, et le mot est faible, à tous les membres de l’équipe et des équipes voisines
que j’ai eu la chance de côtoyer au cours de ces trois années, qui ont partagé mon quotidien au
laboratoire et qui ont su être là, dans les bons moments comme dans les plus difficiles. Leur
écoute, leurs conseils, le partage de leur expérience, leur bonne humeur… tout ceci a
contribué à l’accomplissement de ce travail.

4

Sommaire
5
Sommaire
Avertissements .....................................................................................9
Avant-propos......................................................................................15
Chapitre I: Bibliographie et objectifs..............................................19
I. Les cyclohexa-2,4-diénones ...........................................................21
I.1. Un synthon clé pour la synthèse de molécules naturelles.........................21
I.1.1. Ortho-quinols naturels............................................................................................23
I.1.2. Les ortho-quinols époxydés naturels......................................................................32
I.1.3. Addition nucléophile sur des cyclohexa-2,4-diénones ...........................................33
I.1.4. Les dimères naturels d’ortho-quinols .....................................................................34
I.2. Voies d’accès aux cyclohexa-2,4-diénones...............................................39
I.2.1. Généralités ..............................................................................................................39
I.2.2. Les réactifs iodés hypervalents...............................................................................41
I.2.3. Applications de la réaction de désaromatisation oxydante de phénols à la synthèse
de molécules naturelles ....................................................................................................50
II. Présentation des objectifs ............................................................67
Chapitre II: Résultats........................................................................69
I. La réaction HPD pour la synthèse totale de l’aquayamycine ...71
I.1. Présentation générale.................................................................................71
I.2. Etudes préliminaires..................................................................................76
I.3. Stratégie de synthèse .................................................................................78
I.4. Vers la synthèse de l’aquayamycine .........................................................79
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
1
/
242
100%