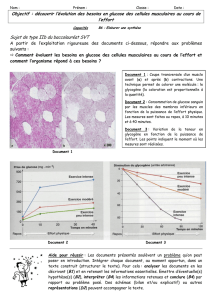
01.04.2015 THESE Mohamed-Sami TRABELSI

1
DOCTEUR DE L’UNIVERSITE DE LILLE 2
Rôle du récepteur nucléaire FXR dans la régulation de
la production de GLP-1 : nouvelle cible thérapeutique
dans le traitement du diabète de type 2 ?
UNIVERSITE LILLE 2 DROIT ET SANTE
Faculté des Sciences Pharmaceutiques et Biologiques
THESE
pour l’obtention du grade de
Unité INSERM U1011
Université Lille 2 Droit et Santé – Institut Pasteur de Lille
Présentée par
Mohamed-Sami TRABELSI
Devant le jury composé de:
Discipline : Aspects Moléculaires et Cellulaires de la Biologie
Spécialité : Biochimie, Physiologie et Biologie Cellulaire
Soutenue publiquement le
1er avril 2015
Président du jury
Rapporteur
Rapporteur
Examinateur
Directrice de thèse
Pr. Bart STAELS
Pr. Nathalie DELZENNE
Pr. Christophe MAGNAN
Dr. Xavier COLLET
Pr. Sophie LESTAVEL

2

3
REMERCIEMENTS

4
Je tiens à remercier le Professeur Bart STAELS pour m’avoir accueilli au sein de son
unité pendant ses quatre ans et demi (en comptant le Master 2) ainsi que pour l’aide qu’il
m’a apportée.
Je remercie Madame le Professeur Nathalie DELZENNE et Monsieur le Professeur
Christophe MAGNAN d’avoir accepter d’être les rapporteurs de mon travail de thèse.
Je tiens aussi à remercier le Docteur Xavier COLLET pour avoir bien voulu participer
à mon jury de thèse ainsi que pour le temps qu’il m’a consacré lors de ma venue à Toulouse.
Je tiens également à vivement remercier le Professeur Sophie LESTAVEL pour avoir
su faire preuve de patience et de grande pédagogie à mon égard. Merci pour la rigueur et
pour le goût de la recherche que vous m’avez transmis. Sophie, ces années m’ont appris
beaucoup sur moi-même et sur les autres, cela est en grande partie grâce à vous.
Véronique, sache que ton aide et ton écoute m’ont été essentielles pour gérer de
manière plus sereine ces années de doctorat pleines de pression et parfois de désillusions.
Heureusement que tu étais là !
A Olivier, merci pour les instants de travail et de détentes passés ensemble. Ce fut
une joie d’être ton voisin de paillasse ces quelques années.
Camille et Sarah, vous reprenez le flambeau des ‘thésards’ du groupe intestin et je
sais que vous le porterez bien haut. Camille, arrête les Welch, ce n’est pas sain (bon je te
l’accorde, c’est vrai que c’est délicieux). Tu testes quand la tomate ?!. Sarah, les manips
enfermés à Pasteur jusqu’à minuit en train de couper de l’intestin resteront comme un
souvenir mémorable de ma thèse… De plus, les éclats de rire partagés seront encore
longtemps dans mes pensées. Enfin les filles, bon courage et tenez bon !!!
Valeria !!! Pas la peine de trop m’étendre ici, tu sais que tu es une amie. On reste en
contact. Bon courage à toi aussi !
Alors chère Dr. De Paoli… En ta compagnie et celle de Valeria, j’ai vraiment aimé
partager ma thèse. Nos discussions sur le monde et la science (et nos soirées…) furent
parmi les meilleures. A quand la prochaine ? OK, je t’ai poussé à faire du saut à l’élastique,
mais pourquoi veux-tu te venger en m’obligeant à sauter du haut d’un avion ?! (PS. : sache
que c’est quand tu veux :) ). Tu resteras longtemps une amie chère.

5
A Nabil, Sarah, Wahiba, Michal, Racha, Aline… Un grand merci pour m’avoir soutenu
et supporté durant ces années ! A bientôt !
Hélène, ton écoute et tes conseils m’ont également été précieux. Merci !
Je remercie également les Anciens (Michal, Wahiba, Sarah,…) et les doctorants ‘en
cours’ (Maheul, Claire, Guillaume Le Flamand et Yann Le Breton) d’être qui ils sont. Tenez
bon !
Lise et Marianne, je vous aime.
Kalim, Kamel et Rémi, nos soirées bowling furent mémorables. Promis, je m’entraine
et la prochaine fois ce sera à vous de payer la tournée.
Mes frères, je tiens également à vous remercier pour votre soutien inconditionnel.
Tous les bons moments qu’on a partagé ensemble m’ont aidé et m’aiderons à rester qui je
suis.
Enfin, je tenais plus particulièrement à remercier mes parents, sans qui rien n’aurait
été possible. Quoique bateau, cette formule résume bien tous les sacrifices réalisés depuis
mon enfance pour me permettre aujourd’hui d’être l’homme que je suis. Papa, si dès le
départ j’ai bossé autant, c’était pour te ressembler. Je me rappelais des soirs où, moi enfant,
tu rentrais de Belgique à 22h, épuisé pour repartir avant notre réveil. Tu as toujours été là
pour nous et tu l’es encore aujourd’hui. Maman, je t’aime, tu es la prunelle de mes yeux.
Qu’à jamais on reste une famille unie.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
1
/
149
100%