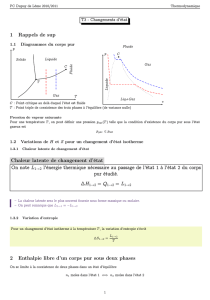
Le corps pur polyphasé

Ch. T3 : Le corps pur polyphasé 26
C H A P I T R E 3
C H A P I T R E 3
LE CORPS PUR POLYPHASE
Dans tout ce chapitre, sauf indication contraire, nous raisonnerons sur un système fermé : l’unité de
masse d’un corps pur.
1. EQUILIBRES ENTRE PHASES DU CORPS PUR
1.1. Variance d’un corps pur polyphasé
L’unité de masse d’un corps pur peut être décrite par trois paramètres intensifs : pression P,
température T, volume massique v.
Lorsque le corps pur est monophasé (état gazeux par exemple), ces trois paramètres sont liés par une
équation d’état du type f(P, v, T) = 0. Il reste alors deux paramètre intensifs indépendants : le système est
divariant, de variance va = 2. Ainsi, à P et T fixées, le volume massique du corps dans l’état considéré
(solide, liquide, vapeur) est fixé.
Quand deux phases coexistent en équilibre, la valeur de la pression impose celle de la température et
réciproquement : il existe donc, entre la pression et la température d’un système diphasé en équilibre,
une relation du type P = f(T). Le système devient monovariant, de variance va = 1 .
On doit remarquer cependant qu’une nouvelle variable intensive précisant l’état du système apparaît :
la proportion relative de chacune des phases du corps pur. Pour l’équilibre liquide-vapeur par exemple,
on définit le titre vapeur x = mv
mT = nv
nT : si x = 0,8, le système est formé de 80% (en masse ou en quantité
de matière) de vapeur et de 20% de liquide. Les fonctions d’état du mélange à l’équilibre (en particulier
le volume massique) dépendent, on le verra, de ce paramètre.
Enfin, le système très particulier formé de trois phases (solide, liquide, vapeur) en équilibre est de
variance nulle, va = 0. Ceci implique qu’il n’existe qu’une pression PT et une température TT où les trois
phases coexistent en équilibre, l’indice T signifiant triple ou triphasé.
1.2. Représentations de l’état d’un corps pur
1.2.1. Surface des états
Aux trois variables P, v, T, on peut associer trois axes orthogonaux et dans cet espace à trois
dimensions, associer un point à l’état d’équilibre d’un système.

Ch. T3 : Le corps pur polyphasé 27
Tous les points d’équilibre d’un système monophasé, liés par f(P, v, T) = 0 seront sur une même
surface caractéristique de l’état, solide, liquide ou vapeur de ce système.
Tous les points d’équilibre d’un système diphasé seront également sur une surface, caractéristique de
l’équilibre (liquide-gaz, solide-liquide, etc...). Les trois surfaces correspondant aux trois équilibres
possibles sont orthogonales au plan (P, T) puisque, dans ce plan, l’équilibre est représenté par une courbe
P = f(T).

Ch. T3 : Le corps pur polyphasé 28
Enfin, tous les points caractéristiques de l’état triple sont sur une droite se projetant en un point (PT,
TT) dans le plan (P, T).
Comme on le voit, même si la représentation à 3 dimensions caractérise le mieux l’état d’un corps pur
(sauf en ce qui concerne l’état triple), des représentations projectives bidimensionnelles, notamment dans
les plans (P, T) et (P, v) sont très utiles à la discussion.
1.2.2. Diagramme (P, T) des états du corps pur
Dans le diagramme (P,T) la dimension de l’espace d’existence d’un système en équilibre correspond à
sa variance : au corps pur monophasé correspond une surface, à un système diphasé une courbe
d’équilibre, au système triphasé un point ( d’où le nom de point triple ).
Chaque courbe d’équilibre du type 1-2 sépare les domaines de stabilité des systèmes monophasés
dans les états 1 et 2. Les trois courbes S-L, S-V, L-V se rencontrent au point triple. En ce point, la pente
de la courbe relative à l’équilibre S-L est très élevée et, en valeur absolue, toujours plus grande que
celles relatives aux équilibres S-V et L-V. D’autre part la pente de la courbe d’équilibre S-V est plus
importante que celle de l’équilibre L-V.
Rq. Les trois pentes des courbes d’équilibre sont positives, sauf dans les cas exceptionnels de l’eau et
du bismuth, pour lesquels la pente de l’équilibre S-L, toujours très élevée, est négative.
La courbe L-V est limitée par le point critique : au-delà de la température critique TC, il n’y a plus de
discontinuité entre l’état liquide et l’état vapeur. Ceci signifie que toutes les grandeurs relatives au corps
(densité , indice optique, constante diélectrique etc...) , qui subissent une discontinuité quand on traverse
une courbe, varient continûment : on parle d’état fluide, ou de fluide hypercritique. Cet état est
représenté par une surface dans le diagramme tridimensionnel, surface limitée par l’isotherme critique
T = TC.
P
T
S
V
L
C
T
TT
TC
PT
PC
etat
fluide

Ch. T3 : Le corps pur polyphasé 29
1.2.3. Diagramme de Clapeyron de l’équilibre L - V
Il fait apparaître trois zones : celles du liquide seul et de la vapeur seule (systèmes monophasés) et
celle de la coexistence du liquide et de la vapeur (système diphasé). Cette dernière zone est limitée vers
le haut par la courbe de saturation, courbe « en cloche » dont le point le plus élevé correspond à la
température TC, et vers le bas par le palier du point triple ( non représenté su la figure ci-dessous ).
P
V
L
PC
L + V
v
vl
vv
(1 - x) v l + x vv
B
M
D
On peut tracer dans ce diagramme différents isothermes (isothermes d’Andrews) qui tous, dans le
domaine liquide-vapeur seront des segments horizontaux reliant le liquide saturant à la vapeur saturante :
quand on parcourt un tel segment DB de gauche à droite, on passe de la première bulle de vapeur (sur la
courbe d’ébullition ) à la dernière goutte de liquide ( sur la courbe de rosée ).
Le titre x en vapeur est tel que, pour un point M quelconque du palier, de volume massique v :
v = (1- x) vl + xvv, d’où x = DM
DB .
Toute autre grandeur caractéristique (énergie interne massique u, enthalpie massique h, entropie
massique s etc...) du mélange L-V en équilibre est calculable à partir de x et des valeurs extrêmes du
palier selon la formule :
g = (1- x) gl + xgv avec x = DM
DB
On peut également faire apparaître dans un tel diagramme les courbes d’isocomposition x = cte. Ce
réseau de courbes permet par exemple de déterminer immédiatement si au cours d’une compression
isochore un mélange liquide-vapeur s’enrichit ou s’appauvrit en vapeur (en fait, si V > Vc le mélange
s’enrichit en vapeur).

Ch. T3 : Le corps pur polyphasé 30
1.2.4. Diagramme entropique de l’équilibre L - V
Ce diagramme associe à l’état d’équilibre du système un point de coordonnées s, T, où s est son
entropie massique. Il fait apparaître les mêmes zones que le diagramme précédent. Le réseau
d’isothermes est simplement remplacé par un réseau d’isobares...
T
V
L
TC
L + V
B
M
D
On a, là encore x = DM
DB
1.3. Utilisation des diagrammes - Pression de vapeur saturante
Considérons un état d’équilibre du système en phase vapeur par exemple ( point A dans les différents
diagrammes ), à la température T0 et la pression P0. Effectuons alors une compression isotherme
réversible : le point représentatif de l’état du système rejoint progressivement la frontière L-V. En B, on
atteint le point où apparaît la première goutte de liquide : on dit que la vapeur est saturante .
La pression d’équilibre L-V à une température donnée est la pression
maximale admissible de vapeur à cette température : on parle de
pression de vapeur saturante souvent notée Ps (T).
Une tentative de compression supplémentaire en ce point ( par diminution du volume par exemple )
ne change pas la pression mais fait progressivement passer le système de l’état vapeur à l’état liquide.
Rq. Ce processus n’existe que pour une température inférieure à TC. D’où l’affirmation :
La température critique est celle au-delà de laquelle on ne pourra
jamais liquéfier une vapeur par simple compression.
Les lois PS(T) sont données pour de nombreux éléments par des lois empiriques parmi lesquelles :
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%