qualification en Médecine Générale Géraldine GUAIS

UNIVERSITE DE NANTES
FACULTE DE MEDECINE
Année 2009 N° 33
THESE
pour le
DIPLOME D’ETAT DE DOCTEUR EN MEDECINE
qualification en Médecine Générale
par
Géraldine GUAIS
née le 20 octobre 1969 à Lille
________
présentée et soutenue publiquement le 19 octobre 2009
Président : Monsieur le Professeur Michel KREMPF
Directeur de thèse : Monsieur le Docteur Patrick GARANDEAU
LE SYNDROME DE BARDET-BIEDL :
UN AUTRE REGARD A L’ILE DE LA REUNION

1

2
Table des matières
JURY 1
I-) INTRODUCTION 6
II-) PRESENTATION DE L’ILE DE LA REUNION 7
II- 1) Situation géographique 7
II- 2) Climat 9
II- 3) La population réunionnaise : un métissage culturel 10
II- 4) Facteurs de consanguinité 11
III-) LE SYNDROME DE BARDET-BIEDL 12
III-1) Historique 12
III-2) Epidémiologie 13
III-3) Diagnostic clinique 14
III-3.1) Rétinopathie pigmentaire 14
III-3.2) Hexadactylie post-axiale et autres anomalies des extrémités 15
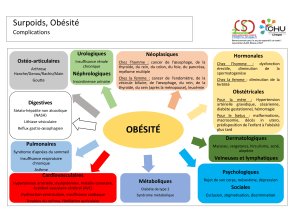
III-3.3) Obésité 16
III-3.4) Hypogénitalisme / hypogonadisme 17
III-3.5) Difficulté d’apprentissage / retard intellectuel 18
III-3.6) Anomalies rénales 18
III-3.7) Autres symptômes mineurs 20
III-4) Diagnostic différentiel 24
III-4.1) Syndrome de Laurence-Moon
III-4.2) Syndrome de McKusick-Kaufmann
III-4.3) Syndrome d’Alström
III-4.4) Syndrome de Biemond II
III-4.5) Syndrome de Prader-Willi
III-5) Génétique 26
III-6) Surveillance et espoirs thérapeutiques 29

3
IV-) MATERIEL ET METHODES 31
IV-1) Recrutement des patients 31
IV-2) Méthodologie 32
V-) RESULTAT 33
V-1) Caractéristiques médicales 33
V-2) Principes de prise en charge 67
VI-) DISCUSSION 68
VII-) CONCLUSION 78
BIBLIOGRAPHIE 79
ANNEXE : Exemples de courbes de corpulence 83
SERMENT 91

4
JURY :
Président du jury :
Monsieur le Professeur Michel KREMPF
Chef de service d’Endocrinologie, Métabolisme et Nutrition,
CHU de Nantes, Hôtel Dieu
Membres du jury :
Monsieur le Professeur Jacques BARRIER
Chef de service de Médecine Interne, CHU de Nantes, Hôtel Dieu
Monsieur le Professeur Jean-Michel ROGEZ
Professeur d’Anatomie, Faculté de Médecine de Nantes
Monsieur le Docteur Cédric LE CAIGNEC
Service de Génétique Médicale, CHU de Nantes, Hôtel Dieu
Directeur de thèse :
Monsieur le Docteur Patrick GARANDEAU
Pédiatre endocrinologue et Chef de service de l’Unité d’Obésité Infantile, Hôpital
d’Enfants, Saint Denis de la Réunion
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
1
/
94
100%