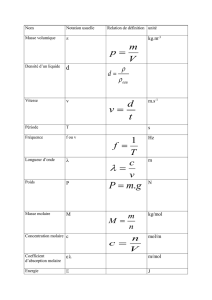
Elimination d`un polluant organique par photodégradation par des

REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE
MINISTER
E
DE L’ENS
E
IGNEMENT SUPERIEUR ET DE LA RECHERCHE
SCIENTIFIQUE
UNIVERS
ITE MENTOURI DE CONSTANTINE
FACULTE DES SCIENCES EXACTES
DEPARTEMENT DE CHIMIE
N° d’ordre
:
Série
:
MEMOIRE
PRESENTE EN VUE DE L’OBTENTION DU DIPLOME DE
MAGISTER EN CHIMIE
ANALYTIQUE ET PHYSIQUE
----------------------------------------
Elimination d’un polluant organique par
photodégradation par des photo
-
inducteurs en
phase homogène et
hétérogène
---------------------------------------
Option
:
E
nvironnement
Par
Zakaria REDOUANE SALAH
Devant le jury
:
Président
: A. ZERTAL Pr Université Mentouri
-
Constantine
Rapporteur
:
M. A.
MALOUKI MC
Université Mentouri
-
Constantine
Examinateur
:
A. BOULKAMH Pr Université Mentouri
-
Constantine
Examinateur : C. MOUATS Pr Université Mentouri
-
Constantine
Souten
ue
le
31/10/2010

A
tou
s
ceux qui me sont
cher
s

Remerciements
Le travail de recherche qui
a
fait l’objet de ce mémoire a été
effectué
en partie dans
le laboratoire
«
Réhabilitation des Ecosystèmes et Développement Durable
»
(REDD) dirigé
par
Madame
Rachida
ASSABAA
, Professeur à
l
’Université
Mentouri
de
Constantine
. Je tiens sincèrement à la remercier pour sa collaboration
gracieuse et la confiance qu’elle m’a accordée en m’accueillan
t à
son laboratoire. Le reste
du
travail a été réalisé au
Laboratoire Sciences
et Technologies de l’Environnem
ent
(LSTE) de
l
’Université
Mentouri
de
Constantine.
Je souhaite exprimer ma sincère reconnaissance à
M
onsieur
M
oulay
A
bderrahm
a
n
e
MALOUKI
,
Maître
de
C
onférences
à l’Université
Mentouri
de
Constantine
et
mon directeur de mémoire, qui a di
rigé et suivi mes travaux qui m’a
fai
t
confiance et m’a
a
pport
é
l’aide nécessaire, tant sur le plan scientifique que moral.
Il m’est particulièrement agréable d’exprimer ma gratitude et ma
reconnaissance
à
Monsieur
A
bdennour
ZERTAL
,
P
rofesseur
à l
’Université
Mentouri
de
Constantine
et
chef de notre équipe de recherche
pour l’aide et la confiance
qu’il m’a accordé en m’accueillant au sein de son équipe et également pour l’honneur
qu’il m’a fait en acceptant de présider le jury de soutenance de ce mémoire.
Mes remerc
iements vont également à
M
es
sieurs
A
bdelaziz
BOULKAMH
et
C
haabane
MOUATS
P
rofesseur
s
à l’Université Mentouri
de Constantine d’avoir accepté
d’examin
er
ce mémoire et d’avoir bien voulu faire partie de ce jury.
Je remercie très chaleureusement tous les membres du laboratoire san
s exception
aucune et en particulier Madame
A
mel
ALLOUI
,
Mademoiselle
Nouzha BOUZIANE
et
Monsieur
A
bdelghani
CHATERBACHE
pour leur attitude personnelle ouverte,
amabilités et amitiés.
En fin, je ne pourrais pas terminer ce propos sans
remercier mes fidèles amis qui
m’ont toujours encouragé et soutenu en particulier
: Mohamed, Hichem,
Hamza,
Bilel,
Yazid
…
et surtout Adel pour son apport précieux lors de tirage de ce mémoire.

TABLE DE
S
MATIERES

I
ntroduction
générale
CHAPITRE I
Etude bibliographique
Partie A
: les colorants
I.1 Historique des colorants
I.2 Généralités sur les colorants
I.3 Classification des colorants
I.3.1 Classification chimique
I.3.1.1 Les colorants azoïques
I.3.1.2 Les colorants triphénylméthanes
I.3.1.3 les colorants indigoïdes
I.3.1.4 les colorants xanthène
I.3.1.5 les colorants anthraquinoniques
I.3.1.6 les colorants phtacyanines
I.3.1.7 les colorants nitrés et nitrosés
I.3.2 Classification tinctoriale
I.3.2.1 les colorants acid
es ou anioniques
I.3.2.2 les colorants basiques ou cationiques
I.3.2.3 les colorants développés ou azoïques insolubles
I.3.2.4 les colorants de cuve
I.3.2.5
les colorants dispersés
I.3.2.6
les colorants réactifs
I.3.2.7
les colorants directs
I.3.2.8
les colorants à mordants
I.4 Toxicités des colorants
I.4.1 Toxicité des colorants azoïques
I.4.2 Toxicité des colorants triphénylméthanes
I.4.3 Toxicité des colorants indigoïdes
1
3
3
3
5
5
5
5
6
6
6
6
7
7
7
7
8
8
8
8
9
9
9
9
10
11
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
1
/
143
100%