La mucoviscidose: un tournant. 1re partie

Quintessenz
쎲Il y a en principe deux concepts thérapeutiques de la mucoviscidose, d’une
part l’action sur les phénomènes pathogénétiques à la base de la maladie, et
donc une stratégie principalement préventive, et de l’autre le traitement rapide
et adéquat des manifestations cliniques, et donc une stratégie principalement
ciblée sur les symptômes.
쎲Le premier groupe comprend les mesures modulant le produit génique ou le
gène lui-même (stimulation d’autres canaux des ions chlore et modification
médicamenteuse du CFTR [cystic fibrosis transmembrane conductance regula-
tor]) ou substituant le gène défectueux. Les options visant le produit génique
CFTR n’en sont qu’aux premiers stades de développement et ne sont donc pas
encore utilisables en pratique clinique.
쎲La stratégie symptomatique a pour but premier de limiter les conséquences
de la maladie. La diminution de l’inflammation, avec le contrôle de la conta-
mination bactérienne des voies respiratoires et du mucus visqueux qui est à
l’origine de la maladie, sont au premier plan.
쎲Malgré le fait que l’amélioration des interventions thérapeutiques a permis
d’atteindre une amélioration impressionnante de la survie des patients atteints
de mucoviscidose au cours de ces dernières décennies, la transplantation pul-
monaire est toujours le seul traitement possible de la pneumopathie de la mu-
coviscidose au stade terminal.
Summary
Cystic fibrosis then and now. Part 1
쎲Two basic principles govern the treatment of cystic fibrosis (CF) of the lung.
The first aims at influencing the pathogenetic basis of the disease, and is thus
a preventive strategy. The second is geared to early and persevering treat-
ment of disease manifestations and is thus a symptom-based strategy.
쎲The first strategy includes interventions on the gene product or the gene
itself. Examples are stimulation of alternative chloride ion channels or modi-
fication of CFTR by drugs. Another option is to replace the gene by gene
therapy. The treatments interacting with the gene product CFTR are still in
the preliminary stages of development and do not yet form part of clinical
management.
쎲The primary aim of the symptom-based strategy is to palliate the conse-
quences of CF of the lung. The most important options are reduction of airway
inflammation, infection control and mobilisation of the highly viscous mucus.
쎲Despite an impressive increase in survival due to better treatment modes
in recent decades, lung transplantation is the only effective treatment option
in end-stage CF of the lung.
CURRICULUM Forum Med Suisse 2006;6:497–500 497
La mucoviscidose: un tournant. 1re partie
Alexander Möllera, Markus Hoferb, Johannes H. Wildhabera, Annette Böhlerc
aPädiatrische Klinik, Abteilung Pneumologie, Kinderspital Zürich
bMedizinische Klinik, Kantonsspital Winterthur
cDepartement Innere Medizin, Abteilung Pneumologie, Universitätsspital Zürich
Vous trouverez les questions à choix multiple concernant cet article à la page 491 ou sur internet sous www.smf-cme.ch.
Introduction
L’espérance de vie des patients atteints de muco-
viscidose (ou «cystic fibrosis», CF) s’est très net-
tement améliorée au cours de ces vingt derniè-
res années. Le diagnostic de mucoviscidose était
la plupart du temps associé à une vie aussi courte
que malheureuse. Mais aujourd’hui les patients
atteints de mucoviscidose ont en grande majorité
une enfance et une vie d’adulte plus ou moins
normale, avec ou sans transplantation pulmo-
naire.
Les raisons de ce meilleur pronostic sont multi-
ples:
– concepts thérapeutiques plus efficaces;
– prise en charge spécialisée par des équipes
pluridisciplinaires;
– meilleure communication et information;
– attitude plus positive des patients.
La mucoviscidose est une maladie complexe qui,
en plus d’une prise en charge médicale spéciali-
sée, demande un suivi compétent dans les ques-
tions physiothérapeutiques, diététiques et socia-
les. Cette prise en charge globale et compétente
n’est possible par une équipe de spécialistes
traitant essentiellement le tableau clinique de la
mucoviscidose et se tenant au courant des tout
derniers développements grâce à une commu-
nication nationale et internationale. En Suisse,
cette communication se fait dans le cadre du
SWGCF (Swiss Working Group for Cystic Fibro-
sis) qui, en tant que groupe de travail indépen-
dant, fait tout pour discuter et coordonner les
questions et problèmes d’ordre médical. L’amé-
lioration du pronostic et l’introduction de nou-
veaux concepts thérapeutiques ont donné davan-
tage d’espoir aux patients et à leur entourage.
Un point important est que les connaissances sur
les mécanismes génétiques et moléculaires de
cette maladie ont ouvert la voie à de nouvelles
options thérapeutiques. Malgré ces connais-
sances, la voie de la recherche à l’application
clinique est longue et difficile. La transplantation
pulmonaire s’est, il est vrai, clairement établie
comme modèle de traitement, mais d’autres op-
tions médicamenteuses restent par contre sou-
vent des théories scientifiques, pour le moment
du moins. Ceci s’explique en partie par le long
chemin que doit suivre un médicament avant de

[1]. Il provoque d’une part une sécrétion accrue
de chlore et d’eau, et de l’autre augmente la fré-
quence des vibrations ciliaires et améliore donc
la clairance mucociliaire. Les premières études
cliniques montrent que cet agoniste du récepteur
P2Y2 est bien toléré, en inhalation tout au moins
[1]. Nous ne savons pas encore si ces «canaux
de remplacement» parviennent à influencer le
déséquilibre sel/eau épithélial dans une mesure
cliniquement significative.
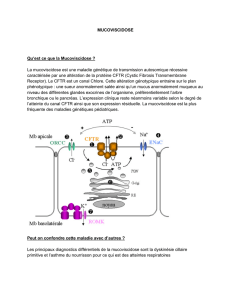
Le CFTR comme cible ou amélioration
de la fonction génique par modulation
du produit génique
La formule structurelle et le mode d’action du
CFTR ont fait l’objet de recherches approfondies.
Ces connaissances détaillées sont exploitées
pour rechercher des options thérapeutiques in-
fluençant la fonction du CFTR, ou permettant son
expression à la surface des cellules. Nous ne sa-
vons toujours pas combien il faut de CFTR pour
obtenir un effet clinique significatif. Il est possi-
ble que 10–35% de l’activité du canal des ions
chlore suffise déjà [2].
Environ 69% des allèles de la mucoviscidose ont
la mutation deltaF508 CFTR et pratiquement
90% des patients sont porteurs d’au moins une
copie de ce deltaF508 CFTR. Cette mutation
donne une protéine CFTR mal constituée, encore
reconnue à l’intérieur du réticulum endoplas-
mique (RE) par le contrôle de qualité des dits
chaperons de la cellule, captée et dégradée dans
les protéosomes. Les substances diminuant cette
fonction de contrôle, c.-à-d. inhibant la fonction
de ces chaperons, permettent au deltaF508
CFTR de sortir du RE et de parvenir à la surface
de la cellule. Là, le CFTR peut conserver sa fonc-
tion, même si elle est nettement diminuée par sa
structure non optimale. Les inhibiteurs des pom-
pes à calcium du réticulum sarcoplasmique/en-
doplasmique (SERCA) ont un tel effet mobilisa-
teur du deltaF508 CFTR. Les exemples de telles
substances sont le MPB, la thapsigargine ou la
curcumine. La curcumine a eu un grand écho
dans les médias, du fait qu’il s’agit d’un consti-
tuant du curcuma, épice du curry, consommé en
grandes quantités dans le monde entier et donc
bon marché [3]. Une fonction correctrice a pu lui
être démontrée dans des lignées cellulaires et
chez la souris, mais les premiers résultats des
études de phase 1 chez des patients atteints de
mucoviscidose ne montrent malheureusement
aucun effet.
Une autre possibilité de modifier le produit gé-
nique est celle des gènes modificateurs. Cette
partie de la recherche profite très largement du
«gene mapping». Le fait qu’il doit y avoir des
gènes modificateurs est confirmé par la variabi-
lité phénotypique du même génotype. Certains
patients porteurs de la mutation homozygote
classique deltaF508 ont une évolution pulmo-
naire très discrète, alors que d’autres présentent
CURRICULUM Forum Med Suisse 2006;6:497–500 498
pouvoir être utilisé couramment en clinique.
L’accès des patients à ces nouveaux traitements
médicamenteux n’est souvent possible que dans
le cadre d’études. Ces nouveaux traitements sont
en outre souvent très chers et ne sont adminis-
trés qu’au compte-gouttes, et de nombreux pays
n’en disposent même pas du tout.
L’article qui suit présente les nouveaux concepts
thérapeutiques qu’ont apporté les connaissan-
ces en génétique et en biologie moléculaire pour
la mucoviscidose, et lesquels se sont établis en
clinique.
De la recherche à la clinique
Il y a en principe deux concepts thérapeutiques
de la mucoviscidose, d’une part l’action sur les
défauts physiopathologiques à la base de la ma-
ladie, qui représente donc une stratégie princi-
palement préventive, et de l’autre le traitement
rapide et adéquat des manifestations de la ma-
ladie, à savoir l’inflammation et la contamination
bactérienne des voies respiratoires, sans oublier
les sécrétions visqueuses qui en sont à la base.
Action sur les défauts physiopathologiques
à la base de la maladie
Les connaissances scientifiques et la compréhen-
sion du défaut de structure au niveau molécu-
laire et génétique, avec la clarification des méca-
nismes physiopathologiques à l’origine de la
clinique complexe que présentent les patients at-
teints de mucoviscidose, ont permis de dévelop-
per des stratégies thérapeutiques modulant ou
substituant le produit génique ou le gène lui-
même. De telles interventions doivent se faire à
un moment de la vie de ces patients où aucune
lésion irréversible ni aucune contamination bac-
térienne chronique n’ont encore pu s’établir.
Les traitements ciblés sur la problématique de
fond visent à améliorer ou remplacer la fonction
du CFTR (cystic fibrosis transmembrane conduc-
tance regulator) défectueux. Toutes les options
agissant sur le produit génique CFTR n’en sont
qu’à leurs premiers stades de développement. De
nombreuses options efficaces en laboratoire (par
ex. en culture cellulaire) ne sont pas encore de-
venus des médicaments utilisables.
Stimulation d’autres canaux ioniques
L’épithélium respiratoire normal dispose de
deux canaux faisant passer les ions chlore au ni-
veau de la membrane apicale, le premier dépen-
dant de l’AMPc, contrôlé par le complexe pro-
téique CFTR, le second étant un canal «alternatif».
Ces connaissances ont fait rechercher des possi-
bilités de stimuler d’autres canaux du chlore que
le CFTR et de contourner ainsi le canal défec-
tueux. Un médicament candidat est le dénufosol
tétrasodique, INS37217, qui active le récepteur
P2Y2 des les cellules de l’épithélium respiratoire

mination rapide et chronique par des patho-
gènes, surtout Pseudomonas aeruginosa, est un
facteur pronostique important de la progression
de la pneumopathie spécifique de la mucovisci-
dose [6].
Un diagnostic précoce de l’inflammation et de la
contamination des voies respiratoires permet un
meilleur traitement et améliore donc le pronos-
tic. Ce diagnostic est posé par BAL. Cette mé-
thode invasive est limitée dans son indication. Il
est également possible de rechercher la contami-
nation sur un frottis pharyngé d’expectorations.
Les cultures de cet examen ont, il est vrai, une
bonne valeur prédictive, mais il y a plus de ré-
sultats faux négatifs ou faux positifs qu’avec le
BAL.
Les marqueurs spécifiques non invasifs de l’in-
flammation des voies respiratoires peuvent être
dosés dans l’air expiré et le condensé respira-
toire. Mais l’intérêt clinique de ces marqueurs
inflammatoires pour la première évaluation et
pour le contrôle de la maladie n’est pas claire-
ment précisé.
Les sécrétions bronchiques pathologiques dans
leur rhéologie, résultant de l’absence du canal du
chlore, sont à l’origine de la colonisation chroni-
que et de l’infection des voies respiratoires, de
même que de la destruction pulmonaire qui en
résulte.
Traitement de l’inflammation
des voies respiratoires
Plusieurs modalités thérapeutiques anti-inflam-
matoires ont déjà été examinées. Les stéroïdes
oraux ont un bon effet, il est vrai, mais aussi des
effets indésirables significatifs à long terme. Au-
cune efficacité n’a pu être clairement démontrée
jusqu’à maintenant pour les stéroïdes en inhala-
tion, mais il est possible que le mucus visqueux
des voies respiratoires en empêche la résorption.
L’ibuprofène a également un effet positif, mais
ses effets indésirables gastro-intestinaux et la né-
cessité de contrôler la formule sanguine en limi-
tent l’emploi à long terme. Le traitement anti-
inflammatoire le plus prometteur est celui par
macrolides (azithromycine, Zithromax®). Les
macrolides ont une bonne diffusion tissulaire et
atteignent une concentration élevée dans les sé-
crétions bronchiques. En inhibant la production
de mucine, et en plus de leur effet anti-inflam-
matoire, les macrolides améliorent la rhéologie
des expectorations et diminuent les facteurs de
virulence de Pseudomonas aeruginosa. Un trai-
tement par azithromycine améliore à long terme
la fonction pulmonaire [7].
Traitement de la contamination
des voies respiratoires
Si ce sont surtout Staphylococcus aureus et Hae-
mophilus influenzae qui jouent un rôle au début
de la maladie, c’est la contamination chronique
par Pseudomonas aeruginosa qui prend la relève
CURRICULUM Forum Med Suisse 2006;6:497–500 499
une baisse rapide et inexorable de leur fonction
pulmonaire. Nous pensons que des gènes modi-
ficateurs protecteurs pourraient être spécifique-
ment stimulés par des médicaments.
Une autre option est la véritable thérapie géni-
que, dans le but d’introduire un gène CFTR nor-
mal dans les cellules des voies respiratoires et de
permettre ainsi la production d’une protéine
CFTR fonctionnelle. Le concept actuel de la thé-
rapie génique somatique dans la mucoviscidose
se base sur l’observation qu’après introduction
d’ADNc plasmidique exogène, codant pour le
CFTR type sauvage, des cellules épithéliales res-
piratoires prélevées chez des patients atteints de
mucoviscidose ont perdu leur mode de sécrétion
ionique typiquement perturbé dans la mucovis-
cidose et acquis une sécrétion ionique physiolo-
gique. Une expression et une structure correcte
de la protéine CFTR, de même que de son trans-
port cytoplasmique vers la membrane cellulaire,
ont par conséquent pu être démontrés par im-
munohistochimie. Ces cellules ont ainsi pu être
phénotypiquement corrigées. Le transfert de ma-
tériel génique n’est plus un problème insurmon-
table. L’épithélium des voies respiratoires a ce-
pendant toute une série de fonctions de défense,
qui empêchent massivement l’introduction
d’ADN modifié dans la cellule. Ces barrières se
sont développées pendant des millions d’années
pour empêcher l’invasion de pathogènes dans les
poumons. Une autre barrière est celle des sécré-
tions denses à la surface des cellules des voies
respiratoires, qui compliquent encore l’introduc-
tion de vecteurs dans les cellules. Le fait de fran-
chir ces barrières défensives est par conséquent
le plus grand défi, qui n’a pas encore été relevé
de manière satisfaisante [4].
Cellules souches: un thème futur
pour la mucoviscidose aussi?
Une lignée de cellules souches embryonnaires
deltaF508 homozygotes a déjà été réalisée et
caractérisée. Il est d’ailleurs également possible
d’aller chercher des cellules souches adultes
dans la moelle osseuse. Le but est d’isoler et de
cultiver ces cellules pour les modifier génétique-
ment, par exemple en leur implantant un gène
CFTR sain. L’idée est de transfuser ces cellules
aux patients, dans l’espoir qu’elles s’installent
dans les voies respiratoires et les colonisent. Au
cas où il y aurait suffisamment de cellules dans
les voies respiratoires, elles pourraient être à la
base d’une production de cellules ayant un CFTR
à fonction normale [5].
Traitement précoce des manifestations
de la maladie
Chez les nourrissons atteints de mucoviscidose,
le lavage bronchiolo-alvéolaire (BAL) montre
déjà une inflammation bronchique non négligea-
ble. Il y a de même une contamination rapide du
système bronchique par des bactéries. La conta-

viscidus). Cette sécrétion de glycoprotéines, eau,
lipides, sels, macromolécules et débris est hyper-
visqueuse sous l’effet de l’extraction d’eau due
au CFTR. Elle favorise la contamination par bac-
téries et levures, en leur offrant d’une part un
milieu de culture idéal, mais aussi en empêchant
les mécanismes de défense de l’organisme et les
antibiotiques de parvenir aux pathogènes. Il y a
longtemps déjà que la physiothérapie intensive a
tenté d’améliorer la clairance de ces sécrétions
visqueuses dans la mucoviscidose. L’inhalation
de désoxyribunucléase humaine recombinante
(rhDNase; Pulmozyme®) diminue la viscosité des
expectorations, améliore la fonction pulmonaire
d’une partie des patients et semble diminuer la
fréquence des exacerbations pulmonaires. Mais
l’effet à long terme de ce médicament très oné-
reux est décevant [11].
L’amélioration des interventions thérapeutiques
a ralenti significativement la progression de la
pneumopathie mucoviscidose, qui reste toutefois
la cause majeure du décès prématuré des pa-
tients atteints de mucoviscidose. Raison pour la-
quelle la transplantation pulmonaire reste la
seule option thérapeutique de la pneumopathie
mucoviscidose au stade terminal. Il s’agit main-
tenant d’un traitement bien établi. La mortalité
périopératoire a pu être drastiquement abaissée
et les patients ont une qualité de vie significati-
vement meilleure. Les résultats à long terme se
sont nettement améliorés ces dernières années
[12].
Remerciement
Nous remercions le Prof. Martin H. Schöni,
Berne, Président du SWGCF, d’avoir lu le manus-
crit.
CURRICULUM Forum Med Suisse 2006;6:497–500 500
avec l’âge. Ces Pseudomonas ont tendance à for-
mer un biofilm mucoïde qui complique sérieuse-
ment l’éradication. C’est pourquoi un manage-
ment antibactérien efficace précoce est capital
pour l’évolution clinique [8].
Une antibiothérapie prophylactique contre Sta-
phylococcus aureus et Haemophilus influenzae
ne présente aucun avantage sur l’évolution de la
maladie. Un tel traitement fait au contraire que
la contamination par Pseudomonas aeruginosa
est plus précoce. Le standard par contre est un
traitement précoce, agressif, en fonction des ré-
sistances, d’une exacerbation infectieuse. Pour
traiter efficacement les infections respiratoires
bactériennes, il faut de très hautes doses d’anti-
biotiques. Ceci s’explique d’une part par la dé-
gradation plus importante de ces médicaments
dans la mucoviscidose, et de l’autre par la diffi-
culté de l’antibiotique à se trouver en concentra-
tions inhibitrices suffisantes dans les pathogènes
en raison de la viscosité des sécrétions. Les an-
tibiotiques en inhalation sont une alternative,
avec l’avantage qu’ils peuvent être délivrés à
hautes doses directement dans les voies respira-
toires [9].
Le diagnostic précoce et l’éradication de la conta-
mination des voies respiratoires par Pseudo-
monas aeruginosa sont difficiles. La vaccination
spécifique par un vaccin conjugué polyvalent dé-
veloppé par Berna Biotech est très prometteuse.
Une vaccination chaque année pendant 10 ans a
donné une diminution des infections à Pseudo-
monas, une amélioration des paramètres de la
fonction pulmonaire et une prise de poids plus
importante des enfants et jeunes adultes traités
[10].
La viscosité de la couche superficielle des voies
respiratoires est à l’origine des manifestations
pulmonaires de la mucoviscidose (lat. mucus
Correspondance:
PD Dr Johannes H. Wildhaber
Pädiatrische Klinik
Abteilung Pneumologie
Kinderspital Zürich
Universitäts-Kinderkliniken
Steinwiesstrasse 75
CH-8032 Zürich
Références
1 Deterding R, Retsch-Bogart G, Milgram L, et al. Safety and
tolerability of denufosol tetrasodium inhalation solution. A
novel P2Y2 receptor agonist: results of a phase 1/phase 2
multicenter study in mild to moderate cystic fibrosis. Pedi-
atr Pulmonol 2005;39:339–48.
2 Kerem E. Pharmacologic therapy for stop mutations: how
much CFTR activity is enough? Curr Opin Pulm Med 2004;
10:547–52.
3 Egan ME, Pearson M, Weiner SA, et al. Curcumin, a major
constituent of turmeric, corrects cystic fibrosis defects. Sci-
ence 2004;304:600–2.
4 Parsons D. Airway gene therapy and cystic fibrosis. J Pae-
diatr Child Health 2005;41:94–6.
5 Wang G, Bunnell BA, Painter RG, et al. Adult stem cells from
bone marrow stroma differentiate into airway epithelial
cells. Potential therapy for cystic fibrosis. PNAS 2005;102:
186–91.
6 Nixon GM, Armstrong DS, Carzino R, et al. Clinical outcome
after early Pseudomonas aeruginosa infection in cystic fi-
brosis. J Pediatr 2001;138:699–704.
7 Dinwiddie R. Anti-inflammatory therapy in cystic fibrosis.
J Cyst Fibros 2005;4:45–8.
8 Ramsey DM, Wozniak DJ. Understanding the control of
Pseudomonas aeruginosa alginate synthesis and the
prospects for management of chronic infections in cystic fi-
brosis. Mol Microbiol 2005;56:309–22.
9 Ramsey BW, Pepe MS, Quan JM, et al. Intermittent admin-
istration of inhaled tobramycin in patients with cystic fibro-
sis. NEJM 1999;340:23–30.
10 Lang ABP, Rudeberg AMD, Schöni MH, et al. Vaccination of
cystic fibrosis patients against Pseudomonas aeruginosa re-
duces the proportion of patients infected and delays time to
infection. Pediatr Infect Dis J 2004;23:504–10.
11 Davis PB. Cystic fibrosis. Pediatr Rev 2001;22:257–64.
12 Boehler A, Weder W. Lungentransplantation – Indikation,
Vorgehen, Chancen und Probleme. Ther Umsch 2005;62:
468–72.
1
/
4
100%










![[tel-00836178, v1] Utilisation des bactériophages](http://s1.studylibfr.com/store/data/005947800_1-ebe8d4fe582003fa637f573dae8589cf-300x300.png)