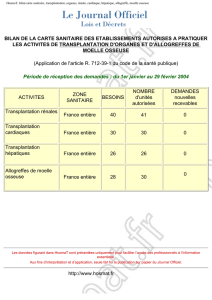
ANNIE BOULANGER FWET DE GREFFES DE MYOBLASTES CHEZ
publicité

ANNIE BOULANGER R ~ L EDES ANTIGÈNES MiNEURS D'HISTOCOMPATIBIUTÉDANS LE FWET DE GREFFES DE MYOBLASTES CHEZ LA SOURIS MDX Mémoire présenté à la Faculté des études supérieures de l'Université Laval pour l'obtention du grade de maître ès sciences (M.Sc.) Microbiologie-Immunologie FACULTÉDE MÉDECINE UNIVERSITÉ LAVAL Mars 1998 O ANNIE BOULANGER, 1998 1+1 National Library ofCanada Bibliothèque nationale du Canada Acquisitions and Bibliographie Services Acquisitions et seMces bibliographiques 395 Wellington Street OttawaON K1AûN4 canada 395, nie WeiIington Ottawa ON K IA ON4 canada The author has granted a nonexclusive licence allowing the National Libmy of Canada to reproduce, loan, distriiute or sell copies of this thesis in microfom, paper or electronic formats. L'auteur a accordé une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distn'buer ou vendre des copies de cette thèse sous la forme de microfiche/nlm, de reproduction sur papier ou sur format électronique. The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts fkom it may be printed or otherwise reproduced without the author's permission. L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation. AVANT PROPOS J'aimerais remercier le Docteur Jacques P. Tremblay pour m'avoir si bien encadrée tout au long de mon projet de recherche. Sa grande disponibilité, sa compréhension et sa confiance furent tout à fait appréciées. De par son enthousiasme, il a su me communiquer une petite parcelle de l'énorme feu sacré qui l'habite en ce qui concerne la recherche. Un gigantesque merci à Isabelle Asselin. C'est elle qui m'a initiée aux rudiments de la recherche. Elle m'a supportée techniquement et moralement à travers ces deux années de maîtrise. Sa patience sans borne et sa coopération ont contribué grandement à la réussite de m o n projet. Merci à Benoit Guérette qui, en me parlant de ses travaux de recherche, m'a donné le goût d'entreprendre ces études. Je le remercie d'avoir omis de me parler des séminaires, clubs de lecture et biopsies sur les bébés souris sans quoi je n'aurais sans doute pas eu le désir de faire cette maîtrise. Je remercie Ikuo Kinoshita pour avoir supporté mes larmes de désolation alors qu'il m'enseignait la culture cellulaire à partir des biopsies de bébés souris. Merci à Jean-Thomas Vilquin pour ses conseils techniques et ses réflexions judicieuses. Il était toujours là, tard le soir, pour discuter boulot ou tout simplement pour me sauver la vie en réparant le cryostat. Merci également à Dominique Mayrand pour ses conseils en biologie moléculaire et à Florence Célestin pour ses encouragements dans les moments difficiles tel l'écriture d'un article... Merci à Brigitte Roy, Marlyne Goulet, Isabelle Deaudelain et François Tardif qui sont toujours là pour résoudre les petits et gros problèmes. Enfin, mille excuses à vous, les petites bêtes, qui nous servez de modèle expérimental. En souhaitant que cela serve à quelque chose de concret et qu'un jour on puisse voir un enfant dystrophique sinon guéri, du moins soulagé! Page &SM ..................................................................................................................... i .. AVANT-PROPOS......................................................................................................... u ... TABLE DES M A T I ~ E S.......................................................................................... LU . LISTE DES TABLEAUX..............., ........................................................... .................vu ... W T E DES FIGURES................................................................... .......................... . .vui LISTE DES FIGURES PRÉSENTÉES D A M L'ARTICLE .................................. ix LISTE DES ABBRÉVIATIONS................... . .......................................................... .x * IÈRE PARTIE ASPECTS FONDAMENTAUX, CLINIQUES ET THERAPEUTIQUESD E LA DYSTROPHIE MUSCULAIRE D E DUCHENNE CHAPITRE 1 LA DYSTROPHIE MUSCULAIRE DE DUCHENNE (DMD) Historique de la maladie.................. ............................................................... 1 Pathophysiologie.............................................................................................. .l Caractérisation de la dystrophine............................................................... 2 1.3.1 Identification du gène en cause dans la DMD et identification du produit du gène................................................................................ 2 1.3.2 Localisation de la dystrophine.......................................................... ..5 1.3.3 Structure de la dystrophine................................................-................5 1.3.4 Distribution de la dystrophine............................................................ 8 1.3.5 Fonctions de la dyçtrophine................................................................ 8 CHAPITRE 2 LA TRANSPLANTATION DE MYOBLASTES COMME AVENUE ~ É R A P E U T I Q U E POUR TRAITER LA DMD 2.1 .................................................... 9 . . La transplantation de myoblastes.......... 2.1.1 Myogénèse et régénération musdaire........ ..... 2.2 ........................ 9 2.1.2 Latransplantationdemyoblastesdiezlasouris ............... . . .....11 2.1.3 Le modèle animal de la DMD: la souris mdx .............................. 2.1.4 Les différents protocoles de transplantations effectués chez la souris par l'équipe du Dr. Tremblay............................................... 14 2.1.5 La transplantation de myoblastes chez l'humain........................16 13 Problème majeur relié à la transplantation de myoblastes .................... 18 CHAPITRE 3 LA RECONNAISSANCE ALLOGÉNIQUE ET LE REJET D E GREm 3.1 . . ........................................................................ 19 La reconnaissance allogeruque 3.2 Le complexe majeur d'hiçtocompatibilité(Ch4.H)....................................... 20 Organisation génétique du CMH...................... . ............................ 20 Rôle du C M . dans la présentation de l'antigène...........................21 Les lymphocytes T: Immunité versus tolérance............................24 Éducation thymique et tolérance naturelle......................................24 . . . ....................................................................................25 L'alloantigenicité Le CMH et la reconnaissance allogénique......................................... 26 Reconnaissance de l'alloantigène par les lymphocytes T..............26 3.3 Répome humorale aux alloantigènes du CMH........................................ -30 3.4 Mécanisme de rejet suivant I'auotransplantation. . ............................ -31 C W I T R E 4 LES ANTIGENES lMINEURS D'HISTOCOMPATISILITE (AGMH) ET LES ANTIGENES NON-ChlH Identifka tion et isolement des AgMH.......................................................... -32 L'antigène mâle H-Y........................................................................................ Différences principales entre CMH et AgMH................................... 33 ...LM Les AgMH sont reconnus uniquement par les lymphocytes T................ 34 Réponse allogénique aux AgMH: Restriction au C M H............................ 35 Assemblage des AgMH avec les molécules du CMH.................................35 Réponse allogénique aux AgMH: Amorçage in vivo ............................... 36 Immunodominance........................................................................................ 36 Expression tissulaire des AgMH.....................................................................36 AgMH et leur implication dans les transplantations.. .... .......................-36 2EME PARTIE ROLE DES ANTIGENES MINEURS D'HISTOCOMPATIBILTTÉ DANS LE REJET D E GREFFES DE MYOBLASTES CHEZ LA SOURIS MDX 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Abstract...............................................................................................................41 Introduction.................................................................................................... ..42 Matenel and rnethods..................................................................................... 43 Results................................................................................................................ 46 Discussion.......................................................................................................... 48 Conclusion..........................,. ...............................................................*............-52 Figures................................................................................................................. 53 References........................................................................................................... 55 3EME PARTIE DISCUSSION GÉNÉRALE . 2. 1 3. 4 5 . Bilan des essais diniques effectués chez l'humain...................................... &4 Retour à I'expérimentation chez l'animal........................ ................. .......65 ............................................................ 66 Importance des AgMH ................... . . Autres études se rapportant au rejet causé par les AgMH .......................... 67 Autres problèmes importants se rapportant à la transplantation de myoblastes................. . 6 ................................................................................... . . 68 Perspectives d 'avenir......................................................................................... 69 LISTE DES OUVRAGES CITES................................................................................ C 71 LISTE DES TABLEAUX Page Table 1: Myoblasts transplantation r e d i s , cellular and humoral reaction of different groups of dogr& and syngeneic g a f t..-..*...*.....*...*...*.........*..****.*.... .-...-.*.-*.-*-..* -.--......-.-...**.*-... *-.* ** * LISTE DES FIGURES Page Figure 1: Localisation du gène de la dystrophine sur le chromosome X ................................................................................... 4 Figure 2: Localisation de la dystrophine par immunohistochimie........... 6 Figure 3: Modèle structural de la dystrophine et de ses protemes associees.............................................................................. 7 8 . 8 Figure 4: Principe de la transplantation de myoblastes.............. . ............ Figure 5: Distribution des cellules satellites dans le muscle strié squelettique............................................................................................ 12 Figure 6: Molécule du CMH de classe I.............................................................. 2 Figure 7: Molécule du CMH de classe II............................................................ 23 Figure 8: Peptides allogéniques présentés par le CMH autologue................28 Figure 9: Peptides allogéniques présenté par le CMH allogénique...............28 Figure 10: Peptides autologues présentés par le CMH dogénique ................29 10 Figure 1: Expression of dystrophin 4 weeks after syngeneic myoblast transplantation Expression of dystrophin 16 weeks after syngeneic myoblast b) transplantation CI, d) A few muscle fibers expressing dytrophin, 16 weeks after myoblast allotransplantation No muscle fiber expressing dystrophin 16 weeks after e) myoblast allotransplantation A few positive fibers expressing dystrophin following O transplantation of male myoblasts in female..................................... 59 a) g) h) i) CD4+ lymphocytes detected 3 to 4 weeks after myoblast allotransplantation MAC+ cells detected 3 to 4 weeks after myoblast allotransplantation CD8+ lymphocytes detected 3 to 4 weeks after myoblaçt allotransplantation.. ............................................................................. .60 Figure 2: Antibodies detected against donor myoblasts..................................61 Figure 3: RT-PCR amplification of granzyme B, y-Interferon and B-actin.................................................................................................... A 2 LISTE DES ABBRÉVU~TIONS DMD: Dystrophie musculaire de Duchenne CMH: Complexe majeur d'histocompatibilité AgMH: Antigènes mineurs d'histocompatibilité Fetal calf serum FCS: HESS: Hank's balanced salt solution INF-11: Gamma- interferon PBS: Phosphate-buffered saline TA: Tibialis antérieur lERE PARTIE: ASPECTS FONDAMENTAUX, CLINIQUES ET THÉRAPEUTIQUES DE LA DYSTROPHIE MUSCULAIRE DE DUCHENNE CHAPITRE 1 LA DYSTROPHIE MUSCULAIRE D E DUCHENNE 1.1 Historique C'est en 1860 que la dystrophie musculaire de Duchenne (DMD) a été décrite pour la première fois par le physiologiste français, Guillaume Ducheme de Boulogne. En 1879, elle fut définie par Duchenne et Gowers comme une myopathie caractérisée par une dégénérescence musculaire progressive accompagnée d'une pseudohypertrophie musculaire. Dans les années 1950, les développements en génétique ont permis de découvrir que cette maladie était liée au chromosome X (Stephem et Tyler, 1951; Walton et Natrass, 1954). Nous avons dû attendre l'avènement de la biologie moléculaire durant les années 1980 pour pouvoir positionner le gène impliqué dans la DMD et identifier la protéine en cause: la dystrophine. 1.2 Pathophysiologie La DMD est donc transmise de mère en fils et affecte environ une naissance mâle sur 3500. C'est la plus fréquente des myopathies et également la plus grave. La DMD est une maladie dégénérative des muscles squelettiques et cardiaques. Elle se traduit par un affaiblissement musculaire progressif affectant successivement les muscles proximaux et distaux. Le phénotype est apparant dès l'âge de 2-3 ans. Malgré la précocité de l'apparition des symptômes cliniques, l'acquisition de la marche est toujours possible. On observe la perte de la fonction ambulatoire vers l'âge de 10-15 ans. Le coeur est souvent atteint et la faiblesse musculaire peut engendrer des déformations osseuses (lordose, scoliose). Un déficit intellectuel est quelque fois observé mais les causes de ce retard mental demeurent inconnues. Le pronostic est mauvais et la mort survient généralement dès le début de la trentaine par déficit cardio-ventilatoire . Parmi les autres observations caractérisant la DMD, la pseudohypertrophie musculaire qui apparaît lorsque les fibres musculaires ne peuvent plus se renouveller à un rythme suffisant pour compenser la dégénérescence. Le muscle s'atrophie alors et les fibres sont remplacées graduellement par du tissu conjonctif et adipeux. Cette prolifération tissulaire entraîne une pseudohypertrophie des muscles. Par ailleurs on retrouve un t a u sérique anormalement élevé d'une enzyme, la créatine-kinase (CK), imputable à la dégénérescence des fibres musculaires. Au point de vue histologique on observe le développement de lésions histopathologiques et ce, dès le début de la myogénèse. Lors de Ia période néonatale on peut voir des modifications minimes, des fibres hypercontractées qui augmentent progressivement ainsi qu'une augmentation de la CK sérique. Quelques mois après la naissance, la fibrose endomysiale apparaît, suivie de la nécrose. A partie de l'âge de 10 ans le tissu fibro-graisseux remplace le tissu musculaire. 1.3 Caractérisation de la dystrophine 1.3.1 Identification du gène en cause dans la DMD et identification du produit de ce gène L'avènement de la biologie moléculaire a rendu possible les recherches en vue d'identifier le gène responsable de la DMD. On savait déjà que le gène en cause était situé sur le chromosome X. Dans les années 80, la découverte de rares filles atteintes de DMD a aidé les chercheurs à positionner le gène. Leurs chromosomes montraient des altérations nommées translocations, c'est-à-dire qu'un bout du chromosome X était échangé avec la partie d'un autosome (chromosomes non-sexuels). Le point de cassure du chromosome X étant toujours situé dans la région p21, il fût suggéré que le gène devait se retrouver à cet endroit. Le processus d'inactivation d'un chromosome X appelé lyonisation peut expliquer le fait que certaines filles soient atteintes de DMD (Lyon, 1961). En effet, tôt lors de I'embryogénèse survient la lyonisation mais puisque les cellules ne peuvent inactiver un autosome, le chromosome X porteur du fragment d'autosome demeure actif, forçant la cellule à inactiver le chromosome normal. D'autres études ont par la suite confirmé que le gène impliqué dans la DMD était bien situé dans la région Xp21 (Murray, 1982, Davies, 1983, Boyd, 1987). La position du gène connue, il restait maintenant à l'identifier. C'est en 1985 que Kunkel et al. ont analysé l'ADN d'un garçon présentant de nombreux symptômes: granulomatose chronique, rétinite pigmentaire, retard mental, syndrôme de McLeod et dystrophie musculaire de Duchenne. En utilisant la technique d'hybridation d e sondes d'ADN complémentaire provenant de la région Xp21, on a pu identifier quatre fragments d'ADN absents chez le malade. Ces sondes ont par la suite été utilisées afin d'identifier le gène en cause. En 1986, le groupe de Monaco et ni ont, par clonage positiomel, réussi à identifier précisément le gène responsable de la dystrophie musculaire de Duchenne. On a par la suite tenté de décoder l'ADN du gène puis identifier la protéine qui en découle, la dystrophine (Hoffman et Kunkel, 1987). Le gène de la dystrophine est le plus grand gène connu avec ses 2,3 millions de nucléotides, soit près de 1% de la longueur totale du chromosome X. Il contient 73 exow aiwi qu'un grand nombre de nucléotides ne participant pas à la synthèse de la protéine ( 0,596 du gène participe à la transcription d'un ARNm de 14 Kb)(Figure 1). Le gène de la dystrophine est retrouvé sur le Figure 1: ciuomosome X au niveau de la région p21. Il est constitué de 2x106 paires de bases et contient 73 exons qui sont transcrits en un ARNm de 14 Kb. 1.3.2 Localisation de la dystrophine De façon à localiser la dystrophine, Hoffman et al (1989) a utilisé des polypeptides synthétisés à partir de deux larges régions dlADNc provenant du gène de la DMD des souriç mdx (modèle animal de la DMD). Pour ce faire, ils ont hybridé ces fragments avec un gène d'E. coli puis incorporé le tout à l'intérieur des bactéries. Les protéines correspondantes obtenues ont été utilisées pour immuniser des animaux. Les anticorps produits ont servi, après purification et sélection pour leur spécificité, à localiser la dystrophine par immunohistochimie (Hoffman et al, 1987-88). Ils ont ainsi pu déterminer sa présence au niveau d u sarcolemme, dans les muscles striés squelettiques des sujets normaux. Par contre, ils ont constaté l'absence de dystrophine dans les muscles des patients souffrant de myopathie de Duchenne ainsi que chez la souris mdx. D'autres auteurs ont également confirmé ces résultats (Monaco et al, 1986; Chelley et al, 1988)(Figure 2) - 1.3.3 Structure de la dystrophine La dystophine est une énorme protéine de 427 kDa contenant 3600 acides aminés. Elle est codée par un gène de 2,5 millions de bases et traduite par un ARNm de 14 kb. Elle représente 0,002% des protéines totales du muscle squelettique et 5% des protéines liées au sarcolemme. C'est une protéine du cytosquelette et elle possède d e nombreuses homologies de structure avec la spectrine et l'a-actinine. La dystrophine est formée de 4 domaines distincts (Koenig et al, 1988): La région N-terminale fixant l'actine est constituée de 200-240 résidus aminés, la partie centrale contituée de 26 séquences en triple hélice, une troisième région riche en résidus cystéine et située entre les résidus aminés 3080-3360 et une extrémité C-terminale contenant 420 acides aminés Ce domaine est lié au complexe glycoprotéique (dystophin-associated glycoprotein complex ou DAG) qui sert d'attache à la face interne d u sarcolemme (Figure 3). Figure 2: Localisation de la dystrophine par immunohistochimie sur des coupes musculaires provenant d'une souris normale (A) et d'une souris dystrophique mdx (B). Figure 3: Modèle structural de la dystrophine et de ses prot6ines associées. La dystrophine forme un complexe glycoprotéique avec six autres protéines membranaires, de façon à Lier la membrane musculaire au cytosquelette interne des fibres musculaires (A). L'absence de dystrophine et de certaines de ses protéines associées déstabilise le complexe et entraîne la vulnérabilité accrue des fibres musculaires dystrophiques (B). Tirée de Enrasti et coll. 1991 (A). Tirée de Hoffman. E.P. La recherche 1993, vol. 24 (B). Normale Dystrophique 1.3.4 Distribution de la dystrophine Chez l'adulte, on peut distinguer deux isoformes: une de type musculaire et une de type nerveuse. Une troisième isoforme dite de type hépatique, de plus petit poids moléculaire, est largement répandue dans tous les tissus. La dystrophine est donc retrouvée dans tous les types de muscles: striés squelettiques, lisses et cardiaques ainsi qu'au niveau du système nerveux. 1.3.5 Fonctions de la dystrophine Le rôle de la dystrophine retrouvée au cerveau est mal compris. Son absence chez les patients DMD pourrait toutefois expliquer le retard mental quelquefois observé. Par contre, la dystrophine d e type musculaire semble d'une grande importance. Mokri et Engel (1975) ont démontré que la membrane des fibres musculaires des patients DMD était rompue à certains endroits. Différents groupes de recherche (Hoffman et al, 1988; Arahata et al, 1988; Rowland et al, 1988) ont permis de localiser par immunofluorescence, la dystrophine sous la membrane plasmatique. Par la suite, Watkins et al. (1988) ont observé par microscopie électronique que la protéine faisait vraisemblablement partie d'un réseau intramembranaire. En 1989, Campbell et al ont démontré que la dystrophine appartenait à un complexe glycoprotéique associé à la membrane. Son rôle principal serait donc d'ordre mécanique et permettrait de renforcir la rigidité du sarcolemme en servant d e lien entre ce dernier, le cytosquelette cellulaire et la matrice extracellulaire. Le déficit en dystrophine, n'empêchant pas la fonction contractile d u muscle, serait mieux perçu comme un facteur déclenchant du phénomène dystrophique que comme un responsable direct. L'intégrité des fibres musculaires dépourvues de dystrophine est grandement compromise. En effet, elles deviennent plus vulnérables aux différentes conditions stressantes, ce qui peut entraîner des perturbations de la perméabilité membranaire, desquelles découlent la nécrose suivie d'une fibrose cellulaire progressive. CHAPITRE 2 LA TRANSPIANTATION DE MYOBLASTES COMME AVENUE THÉRAPEUTIQUE POUR TRAITER LA DMD L'histoire naturelle de la myopathie de Duchenne débute donc par une mutation génétique qui entraîne un déficit en dystrophine. Il s'ensuit un état pathologique déficitaire musculaire progressif accompagné de différentes manifestations histologiques de dégénérescence/régénération. Un mécanisme de régénération inadapté produit une fibrose progressive. Conséquemment, différentes approches thérapeutiques peuvent être envisagées soit en remplaçant directement la dystrophine absente ou en corrigeant l'anomalie génétique par thérapie génique ou encore par transplantation de myoblastes normaux. Enfin, de façon plus modeste, en tentant de contre-balancer le déficit en dystrophine minimisant ainsi le processus de nécrose cellulaire. Dans le présent mémoire, nous discuterons uniquement de la transplantation de myoblastes. 2.1 La transplantation de myoblastes Il s'agit de remplacer la dystrophine absente ou altérée en greffant des cellules précurseurs saines, les myoblastes, qui délivrent le gène normal codant pour la dystrophine fonctionnelle. Cette thérapie cellulaire est basée sur les travaux initiaux de Partridge et al (1989) qui a démontré le pouvoir régénérateur des myoblastes. Ils sont en effet capables de se multiplier et de f u i o ~ e entre r eux ou avec les fibres musculaires de l'hôte pour former des cellules musculaires multinucléées hybrides. Ces dernières sont, par la suite, capables d'exprimer la dystrophine (Figure4). 2.1.1 Myogénèse et régénération musculaire Les muscles sont constitués d'un regroupement de fibres musculaires striées et multinucléées, formées pendant le développement e m b r y o ~ a i r epar la fusion de myoblastes mononucléés. Ces fibres striées sont des structures complexes, hautement différenciées et non-mitotiques. Figure 4: Principe de la transplantation de myoblastes. Cette technique permet de remplacer la dystrophine absente en injectant des cellules saines dans le muscle dystrophique. Les cellules myoblastiques sont obtenues par culture cellulaire suite à la digestion enzymatique des fibres musculaires. n est possible d'obtenir 60 000 myoblastes par gramme de muscle. Par la suite, l'injection de ces cellules au sein des muscles permet d'obtenir des fibres musculaires hybrides contenant des noyaux normaux et des n o y a u dystrophiques. Tirée de Médecine Sciences 1991; 7: 821-829. Un faible pourcentage des myoblastes embryonnaires ne fusionnent pas lors du développement embryonnaire et tombent en quiescence, formant ainsi des cellules satellites en périphérie des fibres musculaires (Figure 5). Ces dernières peuvent être activées lors du bris de la fibre musculaire. C'est alors qu'elles se différencient de nouveau et redeviennent des myoblastes ayant la capacité de se diviser et de fusionner pour réparer la fibre brisée. C'est donc dire que la fusion myoblastique se produit lors de la myogénèse et égaiement pendant la régénération musculaire. Lorsque le bris cellulaire survient, les cellules satellites entrent dans une phase de prolifération intensive. O n ignore encore la nature du stimulus qui déclenche la prolifération des cellules satellites. Il semble toutefois que l'information s'exprime à distance c'est-à-dire au-delà du site de bris cellulaire (Pariridge et Sloper, 1977; Watt el al, 1987). Ce processus est à la base de la transplantation de myoblastes. 2.1.2 La transplantation de myoblastes chez la souris Les premières transplantations de myoblastes chez la souris ont été effectuées en 1988 par Peter Law et son équipe. Ils ont greffé des myoblastes dans le soléaire des souris dystrophiques C57BL/6J @ Y / ~ Y ) Cette lignée de souris sert uniquement de modèle phénotypique pour la DMD car la mutation qui la rend dystrophique n'est siîuée ni sur le gène de la dystrophine ni même sur le chromosome X. Quoi qu'il en soit, Law a constaté que 6 mois après I'injection des cellules, le muscle greffé contenait plus de fibres à l'aspect normal que le muscle témoin contralatéral. Les paramètres observés étaient I'augmentation de la masse, le nombre de fibres ainsi que le potentiel de repos, c'est-à-dire la différence de potentiel entre l'intérieur et l'extérieur des fibres musculaires. Une augmentation significative de tous ces paramètres a permis d e conclure qu'il existait un plus grand pourcentage de fibres saines dans le muscle greffé par rapport au muscle témoin. Par ailleurs, ils ont pu observer une augmentation de la force de contraction isométrique maximale et de la tension tétanique. Selon Law, deux mécanismes pouvaient expliquer ce succès. D'abord, les myoblastes normaux du donneur pouvaient fusionner pour former des myotubes normaux ayant la capacité de remplacer les fibres dys trophiques. Figure 5: Distribution des cellules satellites. Les cellules satefites sont situées sous Ia membrane sarcoplasmique des fibres musculaires (flèche). On peut voir une cellule satellite en coupe longitudinale (A) et transversale (B). Les lettres A, B et C font correspondre les fibres musculaires en A et en B. Ensuite, les myoblastes normaux pouvaient fusionner avec les myoblastes déficients a . de former des fibres musculaires hybrides capables d'exprimer la protéine absente chez la souris dystrophique. Le produit du gène s'avérait donc être le facteur manquant puisque sa présence permettait de corriger le phénotype de la dystrophie. Par la suite, la découverte d'un modèle pour la DMD, la souris mdx, a permis à Partridge et al (1989) de démontrer que suite à l'injection de myoblastes normaux dans les muscles de ces souris, on pouvait observer l'apparition de dystrophhe sous la membrane des fibres musculaires. D'autres travaux ont permis d'établir que même en détruisant le potentiel de cellules satellites de l'hôte par irradiation, il était possible d'obtenir, après transplantation de myoblastes, une régénération quasi complète du muscle chez la souris et chez le chien. Par ailleurs, Morgan (1990) a obtenu 70% de fibres musculaires exprimant de la dystrophine suite à la greffe de myoblastes normaux dans un muscle dystrophique irradié. Ces expériences démontrent bien la capacité qu'ont les myoblastes transplantés à régénérer le muscle hôte. 2.1.3 Le modèle animal de la DMD: la souris m d x Le modèle murin d e la DMD possède une mutation ponctuelle qui remplace un codon d'acide aminé par un codon stop (Sincinski, 1989). Ceci provoque la terminaison prématurée d u polypeptide et se traduit par l'absence de dystrophine dans les muscles de la souris mutante. 11 est important de mentionner l'absence du phénotype caractérisant la pathologie chez les patients atteints de DMD. En effet, la progression de la maladie décrite chez l'humain ne se retrouve pas chez la souris. On observe chez cette dernière un développement normal des muscles, suivie par une myonécrose vers les 20 jours. Par la suite, on remarque une amélioration du muscle du point de vue histologique et la souris ne développe pas la nécrose suivie de fibrose caractéristique à l'humain (Partridge, 1991). C'est pourquoi la souris mdx est un excellent modèle biochimique et génétique de la DMD mais ne peut servir comme modèle phénotypique. 2.1.4 Les différents protocoles de transplantations effectuées chez la souris par l'équipe du Dr. Tremblay Trans~lantationssvneeniaues Les transplantations d e myoblastes histocompatibles pour tous les antigènes d'histocompatibilité (génétiquement identiques à ceux d u receveur) ont conduit à des résultats très intéressants i.e. au-delà d e 90% de fibres muscdaires ayant la capacité d'exprimer la dystrophine, plusieurs semaines après la greffe (Vilquin et al. , 1995). Trans~lantationsalloincornva tibles Les transplantations d e cellules histoincompatibles pour le complexe majeur d'histocompatibilité se sont soldées par un rejet aigu et en aucun temps après la greffe, les fibres n'exprimaient de la dystrophine. L'infiltration lymphocytaire importante et la détection des ARNm de granzyme B et dlIFN-y sont des évidences que cet échec pourrait être imputable aux réactions immunitaires en réponse à la greffe (Guérette et al., 1995). Transdantations sous immunosu~~ression L'équipe du Dr. Tremblay a donc procédé à d'autres transplantations histoincompatibles, cette fois-çi en prenant soin de contrôler la réponse immunitaire du receveur à l'aide d'immunosuppresseurs tels la cyclosporine A (CyA), le FK 506, la rapamycine, la cyclophosphamide et le RS 61443. Parmi les agents irnmunosuppresseurs utilisés, le FK 506 a donné les meilleurs résultats. Lors de transplantations histoincompatibles pour le CMH, l'équipe du Dr. Tremblay a obtenu plus de 80% de fibres musculaires positives pour la dystrophine et/ou P-galactosidase dans les muscles greffés (Kinoshita et al., 1994). Par ailleurs, la plupart des souris traitées au FK 506 n'ont pas développé d'anticorps dirigés contre les myoblastes. Cet agent exerce donc un excellent contrôle sur la réponse immunitaire humorale. Toutefois, le FK 506 n'empêche pas l'infiltration cellulaire (Kinoshita et al., 1994). Le FK 506 a été isolé en 1984, à partir d'une souche de S t r e p t o m y c e s tsukubaenses. Tout comme la cyclosporine, le FK 506 inhibe la production d'interleukine-2 (IL-2)par les lymphocytes T. Ce dernier est cependant de 50100 fois plus efficace que la cydosporine (Kino, 1987). L'efficacité de la rapamycine a également été testée par Vilquin et al. (1995), qui ont démontré que cet agent empêche toujours la formation d'anticorps chez l'hôte et favorise le contrôle du système immunitaire permettant ainsi l'apparition de fibres positives pour la dystrophine dans les muscles greffés. La cyclophosphamide (CPA) a également été étudiée (Vilquin et al.., 1995). Les résultats se sont avérés plutôt médiocres avec peu ou pas de fibres positives pour la P-galactosidase suite aux transplantations sous immunosuppression. La CPA a bien réduit la réponse humorale et on explique ces piètres résultats par l'action toxique de cette drogue qui provoquerait des dommages irréversibles sur les myoblastes injectés. La cyclosporine seule ou en combinaison (CyA+Azathioprine+methylprednisone)n'a pas donné les résultats escomptés. En effet, son efficacité était beaucoup trop variable et on retrouvait des anticorps dirigés contre les myoblastes greffés dans la moitié des cas (Vilquin et al.,, 1994). A utilisée Le RS 61443 s'est avéré complètement inefficace à empêcher la formation d'anticorps (Vilquin et al., 19%). 2.1.5 La transplantation de myoblastes chez l'humain Premières tentatives Les équipes qui ont effectué des transplantations de myoblastes chez l'humain sont au nombre de cinq: celle de P.K.Law ( Memphis, Tennessee, USA), celle de R. Miller ( San Francisco, Californie, USA), celle de G. Karpati (Montréal, Québec, Canada), et celle de J.P. Tremblay (Québec, Québec, Canada). Plus récemment, Mendel et son équipe ont également tenté ce genre de transplantations, mais aucun résultat n'a été publié jusqu'à présent. Nous allons nous attarder plus particulièrement aux travaux effectués par notre équipe. Par la même occasion, nous verrons brièvement les différents protocoles expérimentaux utilisés par les autres équipes. Chacun des protocoles comportent des différences majeures. La sélection du donneur est un des paramètres importants. Les trois premiers groupes ont choisi des individus apparentés aux receveurs tels que le père ou le frère. Le greffon n'était donc pas parfaitement histocompatible. Ils ont évidemment dû employer des immunosuppresseurs tels la cyclosporine A (CyA) et la cyclophosphamide afin de prévenir le rejet. Malheureusement, ces composés ont des effets secondaires importants surtout lorsqu'ils sont utilisés sur une longue période. La CyA est néphrotoxique et à la longue, elle peut être la cause de myopathies (Goy, 1989). Quant à la cyclophosphamide, elle peut entraîner une pancytopénie, une neutropénie ou encore une cystite hémorragique. Chez le docteur Tremblay, des donneurs histocompatibles ont été utiIisés de manière à réduire les risques de rejet immunologique, sans toutefois employer d'immunosuppresseur. Le dormeur était donc compatible avec le receveur pour les antigènes du complexe majeur d'histocompatibilité de classe 1 (HLAA, -B, -C) et de classe II (HLA-Dr). Cette restriction dans le choix des donneurs est nécessaire puisque les myoblastes expriment fortement les HLA de classe 1 (Roy, 1990; Hohlfeld, 1990) et peuvent dam certaines circonstances exprimer les HLA de classe II (Roy, 1991; Hohlfeld, 1990). Les femmes hétérozygotes ont pu servir comme donneuses puisque 50% des clones de myoblastes cultivés peuvent exprimer de la dystrophine, ce qui est amplement suffisant. L'âge des receveurs constitue un autre paramètre expérimental important. Les trois premières équipes ont pratiqué les transplantations chez des sujets relativement jeunes, âgés entre 4 et 10 ans. En effet, la transplantation semble être plus efficace chez de jeunes patients. L'equipe du Dr. Tremblay a préféré effectuer les transplantations chez des adolescents déjà passablement atteints plutôt que de risquer une perte de la fonction motrice suite à un éventuel rejet. Il est déjà connu qu'une histocompatibilité parfaite entre donneur et receveur n'écarte pas nécessairement la possibilité d'un rejet immunologique pouvant être causé par la présence d'antigènes non-CMH, ce qui constitue d'ailleurs le sujet principal de ce mémoire. Le choix du muscle injecté diffère également d'une équipe à l'autre. Celle de Dr. Law a opté pour l'extenseur propre du gros orteil et I'extenseur commun des orteils, évitant ainsi les risques de perte de fonction importante. Les premières injections du Dr. Karpati ont été faites dans le biceps bradual. Enfin, les groupe du Dr. Miller et du Dr. Tremblay ont choisi de greffer au niveau du jambier antérieur. De plus, le nombre de myoblastes injectés variait d'une équipe à l'autre, entre 4 et 100 millions selon les protocoles. Nous avons débuté par une première transplantation de 4 millions de myoblastes suivie de deux injections consécutives de 10 et 50 millions de myoblastes. Nous seuls avons répété la greffe plus d'une fois et dans plus d'un muscle. Les résultats obtenus sont encourageants quoique insuffisants. En effet, neuf patients DMD âgés entre 6-16 ans ont été transplantés. 11 a été observé que le transfert de myoblastes permettait de réintroduire le gène sain et que certaines fibres étaient capables d'exprimer de la dystrophine, quelques semaines après la greffe. Cependant, I'expression de dystrophine a diminué en fonction du temps. Il a également été vérifié que la force volontaire du muscle injecté a augmenté de façon transitoire chez les patients plus âgés pour revenir au même niveau que les muscles non-greffés (Huard et al., 1991; Tremblay et al., 1991). 2.2 Problème majeur relié à la transplantation de myoblastes Les résultats préliminaires de transplantation chez le modèle animal et chez l'humain sont un pas important dans la quête d'un traitement pour la DMD. Néanmoins, les différentes études chez l'humain ont démontré un succès plutôt mitigé puisque la dystrophine, lorsqu'elle est exprimée suite à la greffe, l'est uniquement sur une courte période. Les facteurs pouvant être mis en cause sont nombreux: les conditions de culture des myoblastes du donneur et la mort cellulaire au moment de la greffe, la migration limitée des cellules greffées à partir du site d'injection et majoritairement, la haute surveillance du système immunitaire du receveur face aux antigènes retrouvés sur les cellules du donneur. Ce sont les antigènes du complexe majeur d'histocompatibilité (CMH) qui sont d'abord responsables de la reconnaissance puis du rejet immunitaire. Par ailleurs, même dans le cas d'une histocompatibilité parfaite entre le donneur et le receveur, le problème du rejet persiste. Ce sont alors les antigènes mineurs d'histocompatibilité (AgMH) retrouvés sur les cellules greffées qui pourraient être mis en cause et qui feront l'objet du présent mémoire. Les deux systèmes antigéniques (CMH) et (AgMH) seront traités dans les chapîtres 3 et 4. CHAPITRE 3 LA RECONNAISSANCE ALLOGENIQUE ET LE REJET DE GREFFE La transplantation d'organes ou de cellules fait couramment partie des approches thérapeutiques dans les cas de dysfonctions d'organes et de maladies dégénératives chroniques. Les récents développements en techniques chirurgicales, les nouvelles formes d'immunosuppression en plus de l'avance énorme concernant les connaissances en immunobiologie ont permis d'améliorer grandement les succès de greffes. Quoi qu'il en soit, les échecs sont encore possibles et le rejet chronique en demeure l'une des causes principales. Il est donc important de bien connaître la biologie du système immunitaire, l'un des principaux responsables du phénomène de rejet, et ainsi améliorer les chances de survie du greffon. 3.1 La reconnaissance allogénique Le phénomène de la reconnaissance allogénique a été remarqué pour la première fois lors de transplantations de tissus incompatibles. Ainsi, le rejet de greffes allogéniques a mené à l'identification du complexe majeur d'histocompatibilité ( C m )et de ses produits, qui furent d'abord appelés antigènes de transplantation». Le CMH joue un rôle complexe dans la discrimination immunitaire entre les molécules du «soi» et du mon-soi,) et promeut la détection et l'éradication des agents infectieux. Par ailleurs, on sait depuis longtemps que les tissus incompatibles a u niveau du CMH peuvent induire une forte réponse immunitaire primaire in vitro comme il est possible de le voir dans les réactions croisées de lymphocytes. Une des raisons principales pouvant expliquer la vigueur de la réponse allogénique est le haut pourcentage de lymphocytes T étant impliqués dans la reconnaissance des molécules du CMH allogénique. On peut résumer ce phénomène par le fait que les alloantigènes peuvent être présentés de plusieurs manières, recrutant ainsi un plus grand nombre de cellules lors de la réponse allogénique. 3.2 Le complexe majeu d'histocompatibilité La participation obligatoire du complexe majeur d'histocompatibilité lors de la reconnaissance d'un antigène étranger par les lymphocytes T fait de ces molécules le pivot central de la régulation de la réponse immunitaire et ce, à différents niveaux, notamment dans la détermination d e I'immunogénicité moléculaire, dans la sélection d u répertoire des lymphocytes T, dans l'allogénicité ainsi que dans I'autoimmunité. C'est lors de recherches sur le cancer que l'on a découvert le C H . Durant la première partie du 20e siècle, les études sur la biologie du cancer ont permis d'établir cinq lois concernant la transplantation. (1) les isogreffes sont bien acceptées (2) les allogreffes sont rejetées (3) les tissus provenant de la souche parentale et greffés chez un animal de la première génération (FI)sont acceptés (4) les tissus provenant des parents et greffés aux animaux de la deuxième génération (F2) sont acceptés en partie seulement (5) les tissus provenant de F2 ainsi que des générations subséquentes sont bien acceptés par FI (Snell, 1981). Donc comme il était possible de propager des tumeurs uniquement chez des souris génétiquement très apparentées (inbred), il fût proposé que la base de ce phénomène était la reconnaissance immunologique, par le système immunitaire, de molécules associées aux différents tissus. Ces molécules, uniques pour une souche donnée d'un animal, peuvent diriger la r e c o ~ a i s s a n c eet le rejet d'un tissu étranger provenant d e la transplantation chez un receveur génétiquement différent. 3.2.1 Organisation génétique du CMH Le CMH est un ensemble de plus de 40 gènes qui sont regroupés de façon très rapprochée sur le chromosome 6 chez l'humain et 17 chez la souris. Le complexe HLA (Human Leukocyte Antigens) occupe une région de 3500 kb d'ADN chez l'humain et le complexe H-2 pour sa part, occupe 1500 kb d'ADN chez la souris. HLA et H-2 codent pour trois classes de molécules (classe 1, II, III ) Nous verrons les 2 premières classes seulement. Les HLA de classe 1 et II sont des gènes hautement polymorphes, ce qui agrandit considérablement le répertoire immunologique de chaque individu, créant ainsi un haut degré d'histoincompatibilité au niveau d'une population. Les HLA de dasse 1 comprennent HLA-A, -8,-C en plus des non-classiques découverts récemment; HLA-E, -F, -Gr -H et -J. Ils sont constitués d'une chaîne a-polymorphique associée avec une chaîne p2-microglobuline nonpolymorphique. Ces antigènes sont présents sur la plupart des cellules nucléées et sont particulièrement abondants sur les cellules lymphoïdes et sur l'endothélium vasculaire. (Figure 6). Les molécules HLA d e classe II comprennent 6 sous-régions: DN, DM, DO, DP, DQ, DR. Elles sont formées de 2 chaînes a et p, liées ensemble d e façon covalente. Les antigènes de classe II sont fortement exprimés par les cellules présentatrices d'antigènes (CPA), les lymphocytes B, les cellules dendritiques des capillaires et les macrophages, ainsi que par les lymphocytes T activés (Figure 7). L'expression des molécules du CMH sur les cellules endothéliales et sur les macrophages est grandement influençée par les différents médiateurs immunologiques. Plusieurs cytokines induisent les antigènes de classe 1 (IFNa,p, y ) , Iymphotoxines (TNF a et TNF B ). L'induction des antigènes de classe II est plus réduite, étant réservée à I'IFNy. 3.2.2 Rôle du CMH dans la présentation de l'antigène Les molécules de classe 1 et II situées à la surface des cellules peuvent lier les antigènes. Habituellement, la présentation par le CMH de classe II est réservée aux phagocytes professiomels que sont les monocytes/macrophages et les cellules endothéliales. Ces derniers ont la capacité d'endocytoser les protéines exogènes puis de présenter les produits de la protéolyse (petits peptides ) sur la surface cellulaire, en association avec les molécules de dasse II du CMH. Les lymphocytes T-auxiliaires reconnaissent le complexe CMH classe II/peptide via leur récepteur et répondent de façon appropriée. Figure 6: Diagramme représentant la molécule du CMH de classe 1. La niche peptidique est située entre les domaines al et a2,alors que le domaine a3 sert d'ancrage dans la membrane cytoplasmique. La PZmicroglobuline s'associe au complexe de façon noncovalente. Tirée de Mohanakumar, T. 1994. Peptide Bindhg C=lcIt n Membrane Class 1 MHC Figure 7: Diagramme représentant la molécule du CMH de classe II. La niche peptidique est formée par les domaines al et pl alors que les domaines a2 et 82 servent d'ancrage dans la membrane cytoplasmique. Tirée de Mohanakumar, T. 1994. Peptide Bindiag UeR r Membrane CIass II MHC Les protéines endogènes synthétisées à même la cellule sont, quant à elles, transferrées au réticulum endoplasmique où elles s'associent avec la molécule de classe 1 et la f52-microglobuline. Ce complexe est ensuite <<processé»à travers une série de compartiments intracellulaires jusqu'à la surface cellulaire où il sera reconnu par les lymphocytes T-cytotoxiques. La présentation des protéines endogènes n'est pas limitée aux CPA puisque toutes les cellules expriment les CMH de classe I et ont la capacité de présenter l'antigène en association avec les molécules de classe 1. Les CPA ne font toutefois pas la différence entre les protéines d u «soi. et du «non-soi». Ils présenteront n'importe laquelle protéine endogène synthétisée. La discrimination entre le «soi» et le «non-soi» est plutôt l'affaire du lymphocyte T. 3.2.3 Les lymphocytes T: immunité versus tolérance La réponse immunitaire cellulaire semble être exclusivement dominée par les lymphocytes T. Ceux-ci possèdent un complexe moléculaire, le TcR, existant sous deux farmes hétérodimériques, le classique TcR-aB et le plus récemment décrit, le TcR-y6 qui demeure mal défini et fait toujours l'objet d'études. Le TcR-ap permet de reconnaître les molécules de classe I et II du CMH en association avec un peptide. Les récepteurs des antigènes sont générés de façon continuelle par réarrangements aléatoires de segments d'ADN codants pour les parties variables des récepteurs. Ainsi, des récepteurs spécifiques pour les antigènes du mon-soi. et également du «soi. sont obtenus. C'est donc dire que la discrimination par le système immunitaire ne peut être acquise génétiquement et doit être apprise. Les cellules T doivent apprendre la tolérance face à ses propres protéines. 3.2.4 Éducation thymique et tolérance naturelle C'est au niveau du thymus que les lymphocytes T-auxiliaires et T-cytotoxiques subissent la maturation et la sélection au bout desquelles ils deviennent immunocompétents et peuvent reconnaître un antigène en association avec le CMH. La sélection, en ce qui concerne la spécificité du TcR, met en jeu deux mécanismes. Le premier ayant lieu au moment d e l'expression du TcR membranaire est la sélection positive et se fait parmi les cellules T qui réagissent avec le «soi)>.Le «soi» est représente par les molécules de classe 1 et II du CMH exprimées sur les cellules épithéliales, dendritiques et stromales du thymus. Ainsi, cette sélection positive entraîne la prolifération et la différenciation exclusive des clones auto-réactifs. Le second mécanisme est la sélection négative permettant l'élimination des cellules T ayant une trop forte affinité pour les molécules du CMH du «soi» ou bien pour les molécules du CMH du <<soi.en association avec les antigènes du .soi» (tolérance). Ce dernier cas pourrait mener au grave problème que posent les maladies autoimmunes. C'est à cette étape que l'on doit générer le répertoire antigénique des lymphocytes T. Les antigènes d u CMH ont été identifiés comme les antigènes d'histocompatibilité de par l'évidence de leur rôle dans le rejet de greffes allogéniques Un paradoxe en immunologie qui donne lieu à de nombreuses questions: le haut pourcentage de lymphocytes T (1 sur 10) ayant la capacité de reconnaître les alloantigènes de classe I et II du CMH, et ce, indépendamment de peptides étrangers. A première vue, ce fait semble contradictoire avec l'hypothèse connue de la restriction au CMH disant que pour être activés, les lymphocytes doivent reconnaître un antigène étranger dans le contexte d'un même CMH. Des résultats expérimentaux suggèrent que le haut degré de reconnaissance des molécules d u CMH allogénique pourrait signifier la recomaissance d'un CMH étranger, perçu à la façon d'une molécule du «soi>) altérée. En effet, il existe une similitude suffisante entre le C M . allogénique et CMH du soi pour que la molécule allogénique soit reconnue comme «soi + X». De plus il a été démontré qu'un clone de cellules T donné peut avoir deux spécificités: antigène+CMH d u soi et CMH allogénique en absence d'un antigène. C'est donc dire qu'au niveau d'une population, le pool cellulaire est capable de répondre essentiellement à n'importe lequel CMH allogénique. 3.2.6 Le CMH et la reconnaissance allogénique Le CMH joue un rôle complexe dans la discrimination des molécules du soi et celles qui lui sont étrangères. Ainsi, il permet la détection et l'éradication des différents agents infectieux. Des mécanismes immunitaires similaires permettent la reconnaissance des alloantigènes. La compréhension de ces mécanismes est basée sur deux propriétés du CMH. La première est sa capacité de présenter un peptide donné sur la surface cellulaire pour faciliter la discrimination du soi et d u non-soi. La deuxième est l'extrême diversité génétique du CMH, diversité évidente chez chaque individu par l'expression codominante des HLA de classe I et Il[. 3.2.7 Reconnaissance de l'alloantigène par les lymphocytes Par des mécanismes variés, les protéines endogènes et exogènes sont protéolytiquement clivées en fragments peptidiques, de composition qui dépend de la séquence en acides aminés des protéines ainsi que des enzymes protéolytiques disponibles. De ces pepides, seuls quelques-uns possèdent la conformation nécessaire pour lier la molécule de CMH et seuls ces peptides peuvent devenir disponibles pour la reconnaissance cellulaire par les lymphocytes T. Il est estimé qu'approximativement 1%des peptides peuvent s'insérer dans une molécule particulière du CMH de classe 1. Dans la population, chaque individu exprime de 4 à 6 allèles différents du CMH, chacun pouvant lier une sous-classe de peptides différents. C'est donc dire que les molécules de classe 1 et II du CMH peuvent théoriquement présenter environ 6% de tous les peptides provenant des protéines endogènes et exogènes. Apparemment, cela semble offrir un échantillon protéinique suffisant pour la discrimination immunologique des protéines du soi et du non-soi. En général, seuls les peptides qui sont dérivés des protéines autologues et qui sont liés au CMH autologue peuvent être reconnus immunologiquement comme soi. Tout les autres peptides sont par définition les "non-soi" et sont reconnus comme étrangers. Ceci inclut les peptides dérivés des protéines autologues qui ne lient pas le CMH autologue aussi bien que les peptides dérivés des protéines étrangères qui sont liés au CMH autologue. Le mécanisme est d'autant plus complexe si l'on pense que les cellules T s'engagent aussi bien avec les molécules du CMH autologues qu'allogéniques. Donc les cellules présentatrices d'antigènes (CPA) peuvent provenir de l'hôte et de la greffe. Ces faits rendent la réponse immunitaire allogénique beaucoup plus complexe que la réponse à un agent infectieux. En effet, à cause des multiples manières dont un alloantigène peut être présenté, un plus grand nombre de lymphocytes T est impliqué dans la réponse allogénique. En effet, alors qu'un antigène nominal comme la toxine de Clostridium tetani implique 1/50 000 cellules T pour un seul mécanisme de reconnaissance utilisé, la réponse à un alloantigène engage environ 1/2000 cellules T. On peut ainsi expliquer la forte intensité de la réponse immunitaire primaire lors d'un phénomène de rejet. Il existe au moins trois façons pour les lymphocytes T de reconnaître les alloantigènes: (1) les peptides allogéniques dans le contexte d'un CMH autologue, (2) les peptides allogéniques dans le contexte d'un CMH allogénique, (3) les peptides autologues dans le contexte d'un CMH allogénique. (1)Peptides allogéniques /CMH autologue Il existe des protéines d u CMH allogénique provenant du greffon qui demeurent disponibles pour le système immunitaire du receveur. Ces protéines peuvent être traitées par les CPA et les peptides obtenus sont présentés via le CMH autologue de la même façon que les protéines exogènes (Figure 8). (2) Peptides allogéniques/ CMH allogénique Il est clair que les cellules T du receveur peuvent reconnaître les peptides présentés par les CPA du donneur dans le contexte d'un CMH allogénique. Pour les molécules de classe 1 sur les CPA allogéniques, ces peptides sont présumément dérivés des protéines endogènes comme les molécules allogéniques du CMH elles-mêmes (Figure 9). Figure 8: Diagramme démontrant que des peptides provenant des protéines ailogéniques peuvent être présentés aux lymphocytes CD4+ du receveur en association avec ses propres molécules du CMH de classe II. Figure 9: Diagramme démontrant que des peptides provenant des protéines allogéniques endogènes se retrouvent en association avec les molécules du CMH de classe 1 du donneur et sont présentés a u lymphocytes CD$+ du receveur. Tirées de Mohanakumar, T. 1994. l ALLOGENEIC PEPTIDES / AUTOLOGOUS MHC 3 C I I / / & & CEU AiiûGENElC PEPTiDES / AUOGENEIC MHC Figure 10: Diagramme démontrant que des peptides provenant des protéines du receveur sont disponibles pour lier les molécules du CMH de classe II retrouvées au niveau du greffon afin d'être présentés au lymphocytes CD4+. Tirée de Mohanakumar, T. 1994. AUTOLOGOUS PEPTIDES / ALLOGENEIC MHC R RECIPIENT CL>.+ T = E U (3) Peptides autologues/ C m allogénique Cette situation se présente lorsque les peptides provenant des protéines autologues du receveur étant incapables de lier les molécules autologues du CMH se lient aux molécules de dasse I et II situées sur les CPA au niveau du greffon. Ainsi les peptides dérivés des protéines du receveur peuvent être présentés dans une conformation légèrement différente par les molécules d u CMH du donneur et être reconnus comme étrangers (figure 10). 3.3 Réponse humorale aux alloantigènes du CMH Les anticorps anti-CMH se développent en réponse à un contact avec les molécules du CMH étranger, soit par transfusion, grossesse, ou transplantation de tissu. La réponse initiale au CMH étranger se traduit par la production d'IgM. Etant donné que les molécules du CMH sont d'importantes stimulatrices de lymphocytes T, la réponse humorale est rapidement déclenchée. On assiste alors à une expansion clonale des lymphocytes B, ainsi qu'au changement de classe des immunoglobulines (Ig) en IgG ou IgA. Des alloanticorps sont retrouvés chez les receveurs après une transplantation d'organe de façon très variable. La production d'anticorps chez chaque patient peut se situer entre 10% et 80%. Par ailleurs, à la suite d'un rejet de greffe on retrouve des alloanticorps sériques chez 10% des patients seulement. Les raisons de cette grande variabilité sont incertaines mais pourraient être reliées aux stratégies immunosuppressives utilisées pour les receveurs d'organes. Les facteurs qui influencent la réponse humorale aux antigènes du CMH sont: (1) lïmmunogénicité des molécules du CMH et de leurs épitopes, (2) la prévalence du type de classe de CMH retrouvé sur le greffon, (3) la voie d'immunisation, (4) lïmmunocompétence de l'hôte. 3.4 Mécanisme de rejet suivant l'allotransplantation Le rejet immunologique d'une allogreffe est habituellement orchestré par les lymphocytes T qui s'attaquent aux cellules endothéliales et parenchymales portant les antigènes d'histocompatibilité étrangers, par cytotoxicité directe OU bien par libération de cytokines. La réponse cellulaire est initiée par les lymphocytes lorsque le récepteur TcR reconnaît et se lie au complexe peptide/CMH. Cette interaction est nécessaire quoiqu'insuffisante pour l'activation des cellules T et la prolifération clonale. Une autre catégorie de récepteurs, les molécules d'adhésion, doivent également être présentes afin de favoriser le contact CPA/lymphocyte en plus de fournir les signaux complementaires à l'activation des cellules T. Les mécanismes de rejet impliquent deux types cellulaires: les lymphocytes Tcytotoxiques dans la lyse cellulaire et les lymphocytes T-auxiliaires dans la réaction inflammatoire dérivée de la libération de lymphokines. Dans la plupart des cas, on retrouve les deux classes de CMH au niveau du greffon et par le fait même, la double incompatibilité qui provoque l'activation simultanée par les deux types cellulaires. Les cytokines importantes sécrétées par les lymphocytes lors d'un rejet sont IL2, IFN-y et nuF-P. L-2 déclenche la prolifération cellulaire et est également chimiotactique pour les cellules T. L' IFN-y incite les macrophages à devenir plus cytotoxiques et augmente l'expression des antigènes du CMH sur plusieurs types de cellules. Le TNF-B cause la lyse des cellules sans contact cellulaire. Les perforines, incluant le granzyme B, sont des protéines exprimées uniquement par les lymphocytes T-cytotoxiques activés. Ces protéines sont retrouvées à l'intérieur de granules cytoplasmiques et sont libérées par exocytose contre des cellules cibles spécifiques du greffon (Guérette, 1995). Dans le présent projet de recherche, ce sont 1 ' W - y e t le granzyme B qui seront détectés pour confirmer l'activation des cellules immunitaires infiltrantes suite aux greffes. CHAPITRE 4 LES ANTIGENES MINEURS D'HISTOCOMPATIBILTTÉ ANTIGENES NON-CMH ET LES Les antigènes mineurs d'histocompatibilité sont connus depuis le début des transplantations. Ils sont définis comme étant des alloantigènes retrouvés sur la surface cellulaire. Leur appellation implique qu'ils induisent le rejet de greffes pratiquées entre donneurs et receveurs génétiquement identiques pour tous les loci des antigènes du CMH. Malgré leur appellation .mineur., ces antigènes sont capables dans certains cas, de déclencher des réponses immunitaires vigoureuses. La biologie et la génétique de ce système antigénique ne sont pas encore très bien comprises et son existence demeure la bamère principale à certains types de transplantations notamment la greffe de moëlle osseuse. Ce dernier cas donne lieu à la maladie du greffon contre l'hôte (GVHD), pathologie grave et souvent fatale. Les cas cliniques de rejet de greffes et la GVHD rencontrée chez des patients donc le greffon est HLA-compatible sont l'évidence même de I'existence des antigènes mineurs. 4.1 Identification et isolement des gènes codants pour les AgMH. La méthode utilisée par Snell et al. (1976) pour isoler et identifier les gènes des AgMH a été la production de souches de souris congéniques résistantes. Une souche congénique est définie comme une souche identique à une autre souche (inbred) exception faite d'un segment de chromosome introduit par le croisement avec une autre souche non-apparentée. Dans ce cas, le segment de chromosome étranger comporte des gènes codants pour des antigènes d'histocompatibilité différents et on dit que la souche est congénique résistante puisque que les greffes effectuées entre celle-çi et la souche ainbred), de départ seront rejetées. À l'aide de nombreux (<backcrossing», Bailey (1975) a réussi à établir 25 lignées congéniques à l'aide des souches histocompatibles BalB/c et C57BL/6. Ces lignées de souris sont très utiles pour l'étude des AgMH par transplantations tissulaires ou par essais fonctionnels de lymphocytes T. Il est important de comprendre que ces souches ne diffèrent pas par un seul locus mais plutôt par un segment de chromosome qui peut contenir plus d'un gène codant pour plusieurs AgMH (Bailey, 1982). Les estimations théoriques basées sur les études de transplantation et de &reeding» laissent croire qu'il existerait un très grand nombre d'antigènes mineurs, probablement plusieurs centaines. On peut s'attendre à ce que d e w individus non apparentés comportent de nombreuses disparités antigéniques. L'identification des AgMH chez l'humain est difficile car s'il est relativement rare de rencontrer dans la population des individus génétiquement identiques au niveau des CMH, il est encore plus difficile de trouver des individus qui diffèrent pour un seul locus dlAgMH. Donc, dans le cas où d e w individus seraient compatibles pour le CMH tout en comportant des différences multiples au niveau des loci d'AgMH, il est impossible d'étudier le rôle de chacun des locus séparément. 4.2 L'antigène mâle H-Y L'AgMH humain le mieux défini est l'antigène mâle H-Y. Il est facile à étudier puisque seuls les mâles le possèdent. On l'a remarqué lors de transplantations syngéniques de peau provenant de souris mâles et effectuées sur des femelles (Simpson, 1983). Comme pour les autres antigènes d'histocompatibilité, ce sont les lymphocytes T qui sont impliqués dans le rejet cellulaire via les AgMH et il est possible de cultiver ces cellules in vitro. Puisque les anticorps ne réagissent pas avec l'antigène H-Y,ce sont les lymphocytes T auxiliaires et cytotoxiques qui sont utilisés pour l'étude de cet antigène qui ne semble pas démontrer de polymorphisme. Le gène Hya codant pour l'antigène H-Y est situé sur le bras court du chromosome Y de la souris et est intimement lié au gène Tdy impliqué dans la formation des testicules (McLaren,1988). Chez l'humain, le gène codant pour le facteur de détermination des testicules est situé sur le bras court du chromosome Y alors que l'antigène H-Y est retrouvé sur le bras long (Simpson, 1987). 4.3 Différences principales entre CMH et AgMH Les caractéristiques principales des antigènes mineurs qui les différencient des antigènes du CMH sont l'intensité de la réaction de rejet, la capacité à induire la formation d'anticorps et l'organisation génétique. Contrairement aux antigènes du C H , les AgMH sont généralement moins immunogéniques et ne favorisent pas la formation d'anticorps. De plus, les gènes codants pour les AgMH sont situés partout dans le génome (Roopenian, 1992). En référence aux antigènes du CMH, certains auteurs utilisent le terme non-CMH pour qualifier les antigènes mineurs. En fait, cette dernière appellation pourrait avoir une signification plus globale et inclure toute protéine allogénique. 4.4 Les AgMH sont reconnus uniquement par les lymphocytes T Contrairement aux antigènes du CMH qui peuvent être reconnus par les lymphocytes B et T, la réponse envers les AgMH semble être strictement médiée par les cellules T (Loveland, 1986). En fait jusqu'à présent, aucun laboratoire n'a réussi à produire d'anticorps dirigés contre les AgMH (Loveland, 1986; Rammensee, 1983; Simpson, 1986) ni a détecter la production d'anticorps ou à mesurer l'activité des lymphocytes B en réponse aux différents antigènes mineurs. On pense que le problème est causé par des protocoles d'immunisation inadéquats et qu'il serait difficile de produire des anticorps contre les très courts peptides que sont les AgMH. La réponse des lymphocytes T aux AgMH comportent trois aspects importants: (1)la réaction est restreinte au CMH (2) l'amorçage in vivo est nécessaire (3) la réponse semble se faire selon un patron d'immunodominance. 4.5 Réponse allogénique aux AgMH: Restriction au CMH La restriction au C M . dans la réponse des lymphocytes envers les AgMH a été démontrée en 1975 par Gordon. En effet dans le cas de l'antigène mâle relié au chromosome Y (Ag-HY), les lymphocytes d'une souris mâle d'haplotype ~ 2 d sont capables de reconnaître l'Ag-HY seulement s'il est présenté par les cellules présentatrices d'antigènes (CPA) d'une souris mâle également d'haplotype ~ 2 d Ceci . implique que les AgMH sont en fait de courts peptides présentés en association avec la molécules du CMH. Dam cet ordre d'idée, les antigènes mineurs pourraient être définis comme étant des peptides-épitopes provenant de protéines endogènes et présentés dans un contexte de CMH du soi. La discrimination entre les protéines du soi et d u non-soi se fait via le TcR des lymphocytes. De cette façon, les AgMH pourraient être impliqués dans le développement du répertoire des lymphocytes T (Simpson, 1989). 4.6 Assemblage des AgMH avec la molécule de CMH Donc, les AgMH sont présentés aux lymphocytes T en association avec une molécule du CMH. La formation de ce complexe moléculaire est appelée <<présentation d'antigène» et nécessite la dénaturation et l e clivage protéolytique de la protéine. Il existe deux manières différentes de présenter les peptides selon qu'ils proviennent de protéines exogènes ou endogènes. Les AgMH font partie du second type protéinique et proviennent en fait des protéines synthétisées au sein même de la CPA. Ces antigènes sont donc «processés» au niveau du cytosol puis, les courts peptides obtenus de 16 acides aminés ou moins sont transportés au réticulum endoplasmique où ils s'associent à la chaîne lourde du CMH de classe 1. Cette association est nécessaire pour le repliement adéquat de la chaîne lourde, l'association avec la PZ-microglobuline et le transport du complexe vers la surface cellulaire en vue de la présentation aux lymphocytes T cytotoxiques. Ces peptides ne peuvent être obtenus par le traitement des protéines exogènes mais ils peuvent être fournis en introduisant des peptides synthétiques dans le milieu de culture (Townsend, 1989). 4.7 Réponse allogénique aux AgMH: amorqage in vivo La détection in vitro de lymphocytes spécifiques pour un antigène mineur donné requiert une phase d'amorçage in vivo puis une restimulation in vitro. On explique ce fait par la basse fréquence des précurseurs de lymphocytes T spécifiques aux AgMH ( Kaminçky, 1988). 4.8 Immunodominance Les études sur les AgMH ont démontré qu'une panoplie de peptides est retrouvée continuellement à la surface de toutes les cellules. Nous savons par ailleurs que lors de transplantation cellulaire, l'animal est mis en contact avec un grand nombre d'AgMH et que l'on génère des lymphocytes T cytotoxiques spécifiques à un petit nombre seulement de ces antigènes (Wettstein, 1989). On ne détecte pas de réponse pour la majorité des antigènes étrangers (Wettstein, 1986). On croit que le mécanisme d'immunodominance en cause implique, lors de I'assemblage, la compétition des différents peptides pour une molécule de CMH particulière (Roopenian, 1992; Komgold, 1990). 4.9 Expression tissulaire des AgMH Si les A g W proviennent des protéines endogènes, on peut s'attendre à ce que seules les cellules exprimant ces protéines présentent les molécules à Leur surface. Il semble qu'effectivement plusieurs loci dlAgMH soient restreints à un tissu particulier (Steinmuller, 1984). Par exemple, l'antigène H-40 est exprimé uniquement par les lymphocytes B (Forman, 1984). 4.10 AgMH et leur implication dans la transplantation Des transplantations de coeur et de peau entre souris congéniques résistantes ont démontré que (1) une incompatibilité au niveau d'un seul locus d'AgMH suffit pour causer le rejet, (2) le temps de rejet varie énormément d'un locus à un autre, (3) l'immunogénicité de certains AgMH est additive, (4) les fernelies rejettent plus rapidement les greffes que les mâles (Snell,1976; Graff, 1978; De Tolla, 1977). La nécessité de tenir compte de la présence des AgMH lors de transplantations allogéniques a déjà été largement démontrée. Par ailleurs, il devient urgent de travailler sur des protocoles qui soient applicables à l'humain et qui se rapprochent le mieux possible des conditions cliniques retrouvées chez un patient DMD. Heureusement il est possible de trouver un donneur compatible pour les antigènes du CMH (HLA-A, -8,-C et HLA-DR). Par contre, il est difficile voire impossible de déceler les AgMH et donc d'établir une histocompatibilté parfaite. De là réside le problème de la transplantation de myoblastes et de ce fait, l'importance de vérifier rôle des AgMH dans le rejet de greffes. Mon projet de recherche consistera donc à vérifier le rôle des antigènes mineurs d'histocompatibilité dans le rejet de greffes de myoblastes chez la souris mdx. Dans la 2ème partie, il sera question des objectifs poursuivis par ce projet, de la méthodologie utilisée ainsi que des différents résultats obtenus suivis de l'article "Role of non-MHC antigens in the rejection of transplanted myoblasts". ZIEMEPARTIE- RÔLEDES ANTIGÈNES MINEURS D~HISTOCOMPATIBILTTÉ DANS LE REJET D E GREFFES D E MYOBLASTES CHEZ LA SOURIS MDX Le rôle du système immunitaire dans le rejet de greffes tissulaires n'est plus à définir. Les principaux antigènes (CMH) impliqués dans le rejet allogénique sont également bien décrit comme on l'a vu précédemment dans ce mémoire. Par contre, le rôle des antigènes non-CMH incluant les antigènes mineurs d'histocompatibilité (AgMH) est moins bien défini. Ces derniers sont connus pour leur capacité à induire le rejet de tumeurs et de greffes de la peau lors de transplantations parfaitement compatibles pour les antigènes du CMH. Jusqu'à présent, l'implication des AgMH dans le rejet de myoblastes transplantés chez la souris n'a été qu'indirectement étudiée (Wemig, 1995; Grounds, 1991). E n effet, des études ont démontré que des transplantations de myoblastes entre des souches de souris CMH-compatibles donnaient des résultats plutôt limités et les auteurs ont pamellement attribué ces insuccès aux AgMH. Mon projet consistera donc à vérifier le rôle des antigènes mineurs d'histocompatibilité dans le rejet de myoblastes chez la souris. Il devient indispensable de bien comprendre l'implication des AgMH puisque cesderniers ne peuvent être identifiés préalablement à la greffe, comme nous l'avons vu au chapître 4. II est donc impératif de savoir s'ils provoquent des réactions immunitaires chez le receveur. Si tel est le cas, de quels types sontelles? De quelle façon peut-on les prévenir afin de permettre le succès de la greffe? Nous avons donc effectué des transplantations syngéniques de myoblastes ainsi que des transplantations allogéniques de myoblastes dont les donneurs étaient incompatibles pour de nombreux antigènes non-CMH (incluant les antigènes mineurs d'histocompatibilité) dans le Tibialis antérieur de souris mâles Les souris hôtes n'étaient pas dystrophiques C57BL/ 10J mdx/mdx. irnmunosupprimées. Les greffes de myoblastes syngéniques ont mené à la formation d'un haut pourcentage (90%)de fibres positives pour la dystrophine, 16 semaines après la transplantation. Il n'y a pas eu d'évidence de réaction immunitaire cellulaire contre les myoblastes du donneur: i.e. pas d'infiltration de lymphocytes CD4t ou CD8+ et pas d'expression d'ARNm pour le granzyme B ou pour l'EN-y. Les transplantations de myoblastes provenant de domeurs histoincompatibles pour les antigènes non-CMH ont produit une augmentation transitoire de fibres positives pour la dystrophine, 4 semaines après la greffe et ce, pour quelques souches donneuses seulement. Pour ces souches, le nombre de fibres dystrophine-positives était réduit, 16 semaines après la greffe. Il y a eu évidence de réaction immunitaire cellulaire: infiltration cellulaire de lymphocytes CD4t et CD8+ accompagnée d'une augmentation de l'expression d'ARNm pour granzyme B et IFN-y. Les transplantations de myoblastes provenant de mâles C57BL/lOJ +/+ effectuées sur des femelles C57BL/ 10J mdx/mdx ont également mené à des résultats médiocres avec quelques rares fibres positives pour la dystrophine accompagnées de I'infitration de cellules immunitaires déjà observée. Dans ce dernier cas, la réponse immunitaire a été attribuée aux antigènes mineurs reliés au chromosome Y. Finalement, des anticorps dirigés contre le sérum de veau fatal ont été détectés à la suite des transplantations allogéniques et de façon inattendue, dans les transplantations syngéniques. Ceci indique que le milieu de culture pourrait être source d'antigènes et que les myoblastes auraient pu acquérir ces antigènes lors de leur passage en culture. Chez la souris, la présence de ces anticorps ne réduit aucunement le succès obtenu lors d'une première transplantation syngénique. ROLE OF NON-MHC ANTIGENS IN THE REJECTION OF TRANSPLANTED MYOBLASTS Amie Boulanger', Isabelle Asselin1, Raynald Roy2, Jacques P. Tremblay* Centre de recherche en Neurobiologie Hôpital de l'Enfant-Jésus 1401,18e Rue Québec (Qc), Canada, G 4 124 Tel: (418) 649-5593 Fax: (418) 649-5910 e-mail: LabNeuroaVMl.ULAVAL.CA Centre de recherche en Rhumatologie et Immunologie Le Centre Hospitalier de l'Université Laval 2705 boul. Laurier Ste-Foy (Qc), Canada,G1V 4G2 Tel: (418) 654-2772 Corres~ondingauthor: L Footnote: Docteur Jacques P. Tremblay Centre de recherche en Neurobiologie Hôpital de l'Enfant-Jésus 1401,18e Rue Québec (Qc), Canada, GU 124 This work was supported by grants from the Association Française contre les myopathies (AFM)and the Muscular Dystrophy Association of Canada (MDAC). Keywords: Muscular dystrophy, minor antigen, myoblast transplantation, mouse, mdx ABSTRACT Myoblasts obtained from donors histoincompatible for several nonMHC antigens (i.e. induding minor histocompatibiüty antigens) and from syngeneic donors were transplanted without any immunosuppression into the muscles of male dystrophic C57BL/lOJ mdx/mdx mice. Myoblasts from syngeneic mice resulted in the formation of a high percentage of dystrophin positive fibers 16 weeks after the transplantation. There was no evidence of a cellular immune reaction against the donor myoblasts: i.e. no infiltration by CD4 or CD8 lymphocytes and no increased expression of granzyme B and yINF mRNAs, Transplantation of myoblasts obtained from donors histoincompatible only for non-MHC antigens produced a transient increase of dystrophin positive fibers at 4 weeks post-transplantation for some donor strains but not for others. For donor strains which did produce an increase at 4 weeks, the number of dystrophin positive fibers was reduced 16 weeks after the transplantation. There was evidence of a cellular immune reaction: infiltration by CD4 and by CD8 lymphocytes, increased expression of granzyme B and y-INF mRNAs. Transplantation of myoblasts obtained from male C57BL/10J +/+ into female C57BL/lOJ mdx/mdx also led to the presence of only a few dystrophin positive fibers with the same signs of cellular immune reaction. In this later case, the cellular immune response was attributed to the H-Y minor antigens. Finally, antibodies against fetal calf senun were detected following both syngeneic and non syngeneic transplantations indicating that the culture medium may ako be a source of antigens. In mice, the presence of these antibodies against culture medium did not reduce the success of a first syngeneic transplantation. INTRODUCTION Duchenne muscdar dystrophy (DMD) is a myopathy caused by mutations in the dystrophin gene located on the short a m of the X chromosome (1,2). This devastating disease affects about 1 in 3500 male live birthç. The lack of dystrophin, which is thought to play a role in maintainhg plasma membrane integrity and stability (3-5), resdts in progressive weakness leading to death by the third decade. Myoblast transplantation is a possible treatment to restore dystrophin expression in the muscle of Duchenne patients and mdx mice (6,7). Several in vivo and in vitro experirnents have shown that donor myoblasts can fuse with one another and with host myoblasts to form muscle fibers expressing dystrophin (8-11). Our researdi group has transpianted MHC compatible myoblasts in DMD patients without any immunosuppressive heatment. These transplantations resulted in art increase of dystrophin positive fibers only in a few patients (12,13,15). One of these patients who formed dystrophin positive fibers in one muscle, showed significant increase of strength which was, however, lost progressively with tirne. This lost of strength was attributed at the time to the presence of antibodies against dystrophin which would damage the dystrophin positive fibers formed by the transplanted myoblasts. Since then, our researdi group has also demonstrated successfu1 myoblast transplantation between syngeneic mice without the need for irnmunosuppressive treatment (11). Abundant (90%) dystrop hin positive fibers were observed in theçe host m d x rnice nine mon* after the transplantation even though antibodies against dystrophin were detected in the host as soon as one month after the transplantation. This recent result clearly indicates that antibodies against dystrophin do not damage the new or hybnd muscle fibers resulting from normal myoblast transplantation. Thus, antibodies agauist dystrophin do not appear to be responsible for the progressive loss of strength previously gained by myoblast transplantation in the DMD patients. An alternative explanation is that donor and host were perfectly compatible in mouse syngeneic transplantations, not only for MHC but &O for au non-MHC antigens, whereas most donors and recipients in the chical trials (12,13,15), although compatible for MHC, differed for one or more nonMHC antigens. These non-MHC antigens include minor antigens (MiHA) which are short peptide epitopes of endogenously synthesized proteins presented by the MHC (16,17,19). Although there are no humoral immune responses against these MiHA, they do trigger cellular immune responses. The term non-MHC antigens is used in the present article to group not only h&HA but also all other non-MHC antigens which could trigger a humoral immune response. These non-MHC antigens could have been responsible for the rejection of both donor myoblasts and muscle fibers formed in part by the donor myoblasts, and could thus have been responsible for the progressive lost of strength previously gained following myoblast transplantation in one DMD patient (13). Moreover, the fact that immune response seems to deshoy not only donor and hybrid cells but also host cells (bystander damage) could explain this lost of strength (14). This hypothesis led us to investigate the role of non-MHC antigens in myoblast transplantation in mice. 1s it possible that non-MHC antigens are responsible for myoblast rejection as they are for skin transplantation? Our experiments demonstrate that non-MHC antigens can indeed trigger the rejection of trançplanted myoblasts. They also show that the male antigens alone (i.e. rninor antigens associated with the Y chromosome) (lû-21)are sufficient to trigger the rejection of male myoblasts transplanted into female mice. MATERIAL AND lMETHODS Mouse myoblast culture Mouse prirnary myoblast cultures were obtained from newborn skeletal muscle biopsies (22). The mice were killed and the a m s and legs skinned and the muscles were cut h t o small fragments which were dissociated at 37O Cf initially with collagenase (600 N/ml; Sigma, St- Louis, MO) for 1h and with trypsin (0.1% wt/vol; Gibco, Grand Island, NY) for 30 min in HBSS solution (Gibco). The cell suspension was grown in 199 medium (Gibco) suppiemented with 15%of fetal calf senim (FCS)and a mixture of penicillin G (10 000 UI/ml) and streptomycin (10 mg/ml). Ceus were harvested at confluence and frozen in 199 medium containing 15% FCS and 10% DMSO,until grafting. Animal used Mouse myoblast cell cultures were obtained from 7 different groups of H2bhaplotype: C57BL/6J, C578L/lOJ, C3H.SW/Sn, Dl.LP/SN, 129/J, BXSB/Mp J, DW / J. Myoblast transplantations were performed in C578L/10J mdx/mdx which are also of the H-2b haplotype. Mice were purchased kom Jackson hboratories (Bar Harbor, MA) and reproduced in our animal fadities. This work was authorized by Laval University Animal Care Cornmittee and conducted according to Canadian Council rules on animal care. Myoblast transplantations Three weeks or less before transplantation, the left hind legs of the mdx mice were cobalt-irradiated (20 Gy) in order to block host myoblast proliferation. One day before transplantation, the left tibialis anterior (TA) was exposed and injected with 5 j.d of notexin venom (5 pg/ml), which has been shown to trigger muscle fiber degeneration without damaging myoblasts. On the day of transplantation, the ce& were thawed quickly in 199 medium containing 15% FCS, washed three times in HBSS without FCS and then concentrated as pellets. Cell viability was assessed using Trypan blue stauiing and was always higher than 95%. The left TA muscles were exposed and injected in several sites with approximately 5 x 106 cells suspended in 10 of HBSS. Blood and muscle collection Mice were deeply anesthetized with 0.1 ml of a solution containing ketarnine (10 m g / d ) and xylasine (10 mg/ml). Blood was colleded by cardiac puncture and mice were perfused with 0.9% NaCl solution containing heparin (2 UI/ml,Organon Teknika, Toronto). Blood was centrifuged and the serum fraction was kept at -20°C for cytofluorometric analysis. Some grafted musdes were placed in a 30% sucrose solution for 24 h and then embedded in K T (Miles Inc. Elkart, In), frozen in liquid nitrogen and serially sectioned at 8 p using a Zeiss cryostat. Other muscles were placed directly in liquid nitrogen to prevent RNA degradation. Tissues were stored at -80°Cuntil RNA extraction. Immunohistochemical detection of dystrophin Nonspecific binding sites were blocked by incubating the cryostat sections with a mixture of horse (3.3%),rabbit (3.3%) and fetal calf (3.3%) sera in PBS for 1 h. The sections were then incubated with anti-dystrophin polyclonal antibody (R27,Genica Co., Worcester, MA) (1/1000, 1 h). The second antibody was an anti-sheep peroxidase-conjugated (1/100,1 h, Dako). The peroxidase activity was revealed with 3,3' diaminobenzidine (1 mg/ml) and hydrogen peroxyde (0.03%) CD4, CDS, MAC4 immunohistochemistry Nonspecific binding sites on cryostat sections were first blocked with horse s e m (10% in Tris buffered saline, 20 min). The sections were then incubated for 1 h with the first antibody: a rat anti-CD4 antibody (clone GK 1.5; ATCC, Rockville, MD), a rat anti-CD8 (anti-Lyt-2, clone YTS 169, gift from Dr Waldmann, Oxford University, Oxford, U.K.)or a rat anti-MAC4 antibody (clone M1/70, ATCC, Rockville, MD, USA). Endogenous peroxidase activity was then blocked with 0.03% hydrogen peroxide in methanol for 15 min. The sections were then incubated for 30 min with rabbit anti-rat antibody biotin-conjugated (diluted 1/150, Dako, Copenhagen, Denmark) followed by 30 min incubation with streptavidin-peroxidase (1/200, Dako). Peroxidase activity was revealed with 33' diaminobenzidine (1 mg/ml) and hydrogen peroxide (0.03%). Flow cytomehic analysis Flow cytometric analysis was used to deted in the transplanted mouse senun the presence of antibodies against donor myoblasts. Samples of the myoblasts used for the transpIantations were further expanded in culture. They were then irypsinized, washed by centrifugation and resuspended in phosphate buffer solution at a final concentration of 107 ceIls/ml. A 40 jd sarnple of this myoblast suspension was then incubated for 60 min at room temperature with 20 pl of the s e m of one of the transplanted mice. The cells were washed in 4 ml of cold PBS and incubated with 200 ~1 of a goat anti-mouse antibody conjugated with fluorescein-isothiocyanate(FITC) (Coulter Co., Hialeah, Fl., USA). Following three rinses in PBS, the percentage of labelled myoblasts was determinated with a flow cytometer (FACSort, Becton Dickinson) operating at 488 nm. RT-PCR The RNA extraction method and the amplification of granzyme B mRNA were done exactly as recently described by our research group (23). The amplification of y-INF mRNA and p-actin mRNA was described by Guérette et al. (24). RESULTS Myoblasts obtained from newbom male and female C57BL/lOJ +/+ were injected into the Tibialis anterior of nine male mice C57BL/lOJ mdx/mdx. The graft success was then estimated by detecting the presence of dystrophin-positive fibers at 4 and 16 weeks after the transplantation. At 4 weeks, the number of dystrophin expressing fibers was relatively high (50%) (Figure la) and reached 90% by the sixteenth week (Figure lb, Table 1). Myoblasts were also obtained Crom MHC-compatible newborn male and fernale mice (H-2b) (C57BL/6Jf C3H.SW/Sn, DW /J, BXSB/MpJ, l29/J and DW / J) and transplanted in male C57BL/ 10J mdx/mdx. These donors were, however, incompatible with the host at many non-MHC loci. Myoblasts were allotransplanted into 7 to 9 mdx mice for each type of donor mouse. The success of grafting was estimated first at 4 weeks after transplantation. At this time, a few muscle fibers expressed dystrophin (Figures lc, d, Table 1)but, by sixteen weeks after transplantation, only occasional dystrophin expressing fibers were found (Figure le, Table 1). H-Y antieen Myoblasts obtained from male C57E!L/lOJ +/+ mice were trançplanted into female C57BL/10J mdx/mdx mice. When graft success was assessed between 1 and 5 weeks after transplantation, few or no dystrophinpositive fibers were seen (Figure If, Table 1). Detection of antibodies aeainst mafted mvoblasts. Antibodies reacting with cultured donor myoblasts were detected in the s e m of almost all mice (41/44) during the 4 weeks following the graft (Figure 2, Table 1). A surprising observation was that these antibodies were detected in the serum of the host, even following syngeneic transplantations (Figures 2c, d), although at very low titres, 16 weeks after transplantation (Table 1). The antibodies present in the semm of hosts given syngeneic g r a h reacted only with donor myoblasts which had been grown in culture with FCS and not when they had been grown without FCS at any stage. In one experiment, C57BL/lOJ +/+ myoblasts were grown without FCS and transplanted in three males C578L/lOJ mdx/mdx. F o u weeks later, no antibodies reacting with the donor myoblasts were detected in the serum of the recipient mice. Immune cell infiltration In all the muscles transplanted with myoblasts obtauied from strains incompatible for non-MHC antigens, very conspicuous infiltrations of CD4 or CD8 lymphocytes and MAC-1 expressing cells (macrophages, neutrophils) were observed in tissue sections, 4 weeks after grafting (Figures lg, h, i). In contrast, only a few lymphocytes were observed in muscle sections of male m d x mice transplanted with syngeneic myoblasts although many macrophages were observed at these injection sites probably due to muscle damage. Detection of R-actin,eranzvme B and lFNy mRNAs RT-PCR amplifications of granzyme B and y-INF mRNAs were performed on muscles obtained from mice transplanted with myoblasts from C57BL/10J +/+, C3H.SW/Sn or BXSB/MpJ donors. The amplifications were normalized between biopsies by co-amplifymg the mRNA of p-actin. No significant increased expression of granzyme B and y-INF mRNAs were observed at 1,2 and 3 weeks following syngeneic transplantations (Le. C57BL/10J +/+ in C57BL/ 10J mdx/mdx). However, significant increases of these mRNAs were detected 1 and 2 weeks after transplantation in muscles injected with C3H.SW/Sn or BXSB/MpJ myoblasts as compared to the contralateral uninjected muscles (Figures 3a, 3b). These results confirmed that the infiltrating lymphocytes, present following non-MHC compatible myoblast transplantations, were activated. DISCUSSION The term non-MHC antigens is used in this article to designate all antigenç which are not major histocornpatibility antigens (MHC), including the minor histocompatibility antigens (MiHA). These MiHA were initially identified by their ability to induce tumor or skin graft rejections following MHC compatible grafts (16,17). The present study indicates that non-MHC antigens are also responsible for the rejection of myoblasts transplanted in mdx mice. Indeed, excellent transplantation results were obtauied with syngeneic myoblaçts, C578L/lOJ +/+ in male C57BL/ 10Jmdx/mdx, as previously reported by our group (11). As in that previous study, more than 90% of the muscle fibers expressed dystrophin in the transplanted mdx muscle, 16 weeks after transplantation. These dystrophin positive fibers resulted from fusion of the normal (non dystrophie) myoblasts and thus indicated that these cells were not rejected even without immunosuppression. The absence of immune reaction was also indicated by the minimal infiltration of the injected muscles by CD4 or CD8 lymphocytes and by MAC-1 positive cells. However, when MHC compatible myoblasts were obtained from mouse strains other than C57BL/lOJ, differing for non-MHC antigens, the success of transplantation was rather limited. Indeed only a few dystrophin positive fibers were observed 4 weeks post transplantation. Some other groups have obtained similar results with MHC compatible muscle allografts in mice (14,261. However, Watt et al. (1991) have observed proionged survival (up to 105 days) of dlogeneic precursor cells within host muscles (27). This could depend on the host/donor strain combinations used. In some cases, the scarce dysfxophin positive fibers may have been due to a reversion rather than to the transplantation (25). Although these donors had the same MHC haplotype as the host (i.e. H2b), they were expected to differ from the hosts at many non-MHC antigen loci. Some differences were obsenred not only in the number of surviving dystrophin-positive fibers but also in the degree of infiltration by lymphocytes and macrophages between the mice which received myoblasts from the various donor strains. This may have been due to a more or less rapid rejection depending on the immunogenicity of the various non-MHC antigens involved. The presence of macrophages and of lymphocytes 4 and even 16 weeks after the transplantation, supports the hypothesis of an immune reaction. This rejection phenomenon is, however, slower than that observed in MHC incompatible myoblast transplantation which leads to complete rejection in less than 2 weeks (23)- It was initidy surprishg to observe the presence of antibodies against the donor myoblasts even foIlowing syngeneic transplantation since the only antigen which was absent from the host was dystrophin. Dystrophin would be expressed only in a low percentage of mature myoblasts and so could not explain the reaction of the host serum with all donor myoblasts. Moreover, dystrophin being an intracellular protein, would not be accessible to the antibodies in non permeabilized ce&. The absence of reaction of the host s e m with donor rnyoblasts, following transplantation of myoblasts grown without FCS, indicated that the antibodies were generated against FCS components attached to the culture myoblasts. These components remained attached to the myoblasts even after three rinses. Our interpretation of the results is now that myoblasts grown in presence of FCS have some FCS components on them at the time of transplantation which provoke a humoral immune reaction in the host mice. As a result, serum obtained from the grafted animals more than 4 weeks after the transplantation contained antibodies which reacted with these components present on the myoblasts grown in FCS and used for the flow cytometry test. AntiFCS antibodies may be partially responsible for the positive reaction with donor myoblasts of sera obtained after transplantation of MHC compatible myoblasts which differ at non-MHC loci. Some of these antibodies may also be directed against minor antigens other than the FCS components; although minor antigenç are comrnonly defined in terms of cellular response this does not preclude the triggering of a humoral response by allotypic immunogenic proteins, MiHA, present on MHC-compatible but non-syngeneic donor myoblasts or on the muscle fibers formed from these ce&. Finally it should be noted that antibodies against FCS are not responsible for the rejection of the transplanted myoblasts since such antibodies were present following syngeneic transplantation and did not lead to the rejection. The presence of a cellular immune reaction against myoblasts obtained from donors incompatible for non-MHC antigens was clearly demonstrated by the expression of granzyme B and W - y by the lymphocytes infiltrathg the myoblast injected muscles. This cellular immune reaction is probably due to the MiHA. It should be emphasized that no sign of cellular immune reaction was observed fouowing fully histocompatible transplantation. The presence of FCS which did trigger a humoral response in these transplantations did not tngger a cellular immune response. The rejection of myoblasts following MHC compatible but MiIIA incompatible myoblasts will probably be adequately controlled by FK506 immunosuppression since this treatment was effective following MHC incompatible myoblast transplantation (10). Indeed in the transplantations reported in that artide, donors and recipients differed not only for MHC but also for many MiHAs. These MiElAs codd have been presented to the host b u n e system by the host MHC on hybnd myotubes (formed by fusion of donor myoblasts with host myoblasts) and yet the immune response was adequately controlled by FK506. Responses to minor antigens in other transplantation systems are only mediated by T cells. The minor antigens wodd be presented as short peptides (8 to 12 amino acids) by MHC class 1 molecules. Myoblasts have been shown to express MHC class I molecules (28) and are thus capable of presenting directly these minor antigens. This w o d d explain why this type of transplantation is still relatively rapidly rejected even when the host and the donor differ only for minor antigens. The failure of the transplantation of male C57BL/10J +/+ myoblasts in female C57BL/ 10J mdx /mdx mice clearly demonstrated that differences of only a few minor antigens, linked in this case to the Y chromosome, are enough to trigger the rejection process. This observation explains why myoblast transplantation evaluated with probes against the Yantigen had relatively little success (2930). In contrast to the conventional approach of using cultured rnyoblasts, the transplantation of whole and sliced musdes was reported by Fan et al. (1996) to give longer survival (up to one year) (31). This could be attributed to a reduced or delayed exposure of the muscle slices to the immune response which could result in the developpment of tolerance. CONCLUSION Non-MHC antigens, including minor histocompatibility antigens, present on myoblasts are recognized by the host immune system and are responsible for the rejection of myoblast transplantation even when the host and the donor are MHC compatible. This observation probably accounts for the limited success that our research group has obtained when MHC compatible myoblasts were transplanted to Dudienne muscular dystrophy patients without immunosuppression (13). It will therefore be necessary to immunosuppress DMD patients even when they are transplanted with MHC compatible myoblasts. This research is supported by gants from AFM, MDA and MDAC. We wish to thank Dr. Terry A. Partridge for revising this manuscript. FIGURE LEGENDS Figure la: Figure lb: Figures lc,d: Figure le: Figure If: Figures lg,h,i Four weeks after syngeneic myoblast transplantation (C57BL/lOJ +/+) in C57BL/10J mdx/mdx mice, a high percentage of the fibers expressed dystrophin revealed by immunoperoxidase. Sixteen weeks after syngeneic (C5ïBL/lOJ +/+) myoblast transplantation, almost 90% of the musde fibers expressed dystrophin as shown by immunoperoxidase. Four weeks after transplantation of myoblasts from mice incompatible for non-MHC antigens C57BL/6J (c) and from l29/J (d) in mice C57BL/ 10J rndx/rndx mice, there were only a few muscle fibers expressing dystrophin (arrows). Sixteen weeks after the transplantation of 129/J rnyoblasts in C57BL/10J mdx/mdx mice there were no muscle fibers expressing dystrophin. Myoblasts obtained from male (C57BL/ 10J +/+) were hansplanted into female (C57BL/lOJ rndx/mdx.) Only a few positive fibers were present 1 to 5 weeks after such transplantation. The presence of infiltrathg cells was detected by immunoperoxidase in muscle sections 3 to 4 weeks after myoblast allotransplantation, CD4+lymphocytes (g), MAC-1 positive cells, i.e. macrophages and neutrophils (h) and CD8+ lymphocytes (i) were observed. (scale bar: a to f: 4 0 0 ~ g; and h: 2 0 0 ~i:; 100p.m) Figure 2: Antibodies against the donor myoblasts (Le. 129/Jin a and b, C578L/lOJ in c and d), were detected in the senun of the host mice by 80w cytometry. In a and c, preimmune serum and in b and d serum obtained 4 weeks after the transplantation. Positive reaction, i.e. increase of the intensity of l a b e h g was observed in the sera obtained 4 weeks after the transplantation in both cases. Figure 3 RT-PCR amplification of mRNA's of granzyme B y-INF @) and $-actin (c). Significant increases of mRNA were detected 1 and 2 weeks after transplantation in the musdes injected with C3H.SW/Sn or BXSB/MpJ compared with the contralateral uninjected muscles. (a), REFERENCES Kunkel, L.M., Monaco, A.P., Middlesworth, W., Odis, H.D. and Latt, S.A. Specific cloning of DNA fragments absent from the DNA of a male patient with an X chromosome deletion. Proc. Natl. Acad. Sci. USA 1985; 82: 4778-4782. Ray, P.N., BelfaU, B., Duff,C. et al. Cloning of the breakpoint of an X:21 translocation associated with Duchenne muscular dystrophy. Nature 1985; 318: 672-675. McArdle, A., Edwards, R.H.T. and Jackson, M.J. Effects of contractile activity on muscle damage in the dystrophin-deficient mdx mouse. Clinical Science 1991; 80: 367-371. Menke, A. and Jockusch, H. Decreased osmotic stability of dystrophinless muscle celk from the mdx mouse. Nature 1991; 349: 69-71. Sacco, P., Jones, D.A., Dick, J.R.T. and Urbova, G. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. C h . Sci. 1992; 82: 227-236. Partridge, T.A. Myoblast transfer: A possible therapy for inherited myopathies? Muscle and Nerve 1991; 14: 197-212. Morgan, J.E. and Partridge, T.A. Ce11 transplantation and gene therapy in muscular dystrophy. Bio. Assays 1992; 14: 641-645. Partridge, T.A., Morgan, J.E., Coulton, G.R., Hoffman, E.P.and Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to positive by injection of normal myoblasts. Nature 1989; 337: 176-179. Huard, J., Verreault, S., Roy, R., Tremblay, M. and Tremblay, J.P. High efficiency of muscle regeneration after human myoblast clone transplantation in SCID mice. J. Clin. Invest. 1994; 93: 586-599. Kinoshita, I., Vilquin, J.T., Guérette, B., Asselin, I., Roy, R. and Tremblay, J.P. Very efficient myoblast dotransplantation in mice under FK506 immunosuppression. Muscle and Nerve 1994; 17: 1407-1415. Vilquin, J.T., Wagner, E., Kinoshita, I., Roy, R. and Tremblay, J.P. Successful histocompatible myoblast transplantation in dystrophindeficient mdx mouse despite the production of antibodies against dystrophin. J. Cell Biol. 1995; 131: 975-988. Huard, J., Bouchard, J.P., Roy, R. et al. 1992. Human myoblast transplantation: Preliminary results of 4 cases. Muscle and Nerve 1992; 15: 550-560. Tremblay, J.P., Malouin, F., Roy, R., Huard, J., Bouchard, J.P., Satoh, A. and Richards, C.L. Results of a triple b h d d i n i c a l study of myoblaçt transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplantation 1993; 2: 99112. Wemig A., Irintchev A. Bystander damage of host muscle caused by implantation of MHC-compatible myogenic celis. J. Neurol. Sci. 1995; 130: 190-196. Amato, A.A., King, W., Signore, L., Prior, T.W., Mendell, J.R., Kissel, J.T., Sahenk, Z., Besson, S., McAndrew, P.E., Rice, R., Nagaraja, H., Stephens, R., Lantry, L., Morris, G.E. and Burghes, A.H.M. Myoblast transfer in the treatrnent of Duchenne's muscular dystrophy. N. Engl. J. Med. 1995; 333: 832-838. Lindahl, K.F. Minor hktocompatibility antigens. TIG 1991, 7:219-224. Roopenian, D.C.,Davis, A.P., Christianson, G.J. and Mobraaten, L.E. The functional basis of minor histocompatibility loci. The Journal of Unmunology 1993; 151: 4595-4605. 18. Eichwald, E.J. and Silrnser, C.R. Transplant Bull. 1955; 2: 148-149. Goulmy, E., Termijtelen, A., Bradley, B.A., Van Rood, J.J. Alloimmunity to human H-Y. Lancet 1976; 2: 1206 Goulmy, E., Termijelen, A., Bradley, B.A., Van Rood, J.J. Y-antigen killing by T cells of women is restricted by HLA. Nature 1977; 266: 544545. Wang, W., Meadows, L.R., Den Haan, H.M., Sherman, N.E.,Chen, Y., Blockland, E. et al. Human H-Y: a male specific histocompatibility antigen derived f ~ o mthe SMCV protein. Science 1995; 269: 1588-1590. Cossu, G., Zani, B., Coletta, M., Bouchè, M., Pacifia, M. and Molinarol, M. Ln vitro differentiation of satellite cells isolated from normal and dystrophie mammalian muscles, a cornparison with embryonic myogenic cellç. Cell Differentiation 1980; 9: 357-368. Guérette, B., Roy, R., Tremblay et al. hcreased granzyme B mRNA after allouicompatible myoblast transplantation. Transplantation 1995; 60: 1011-1016. Guérette, B., Tremblay, G., Vilquin, J.T. et al. hcreased interferon-y mRNA expression following alloincompatible myoblast transplantation is inhibiteci by FK506. Muscle and Nerve 1995; (in press). Hoffman, E.P., Morgan, J.E.,Watkins, S.C.and Partridge, T.A. Somatic reversion/suppression of the mouse mdx phenotype in vivo. J. Neurol. Sci. 1990; 99: 9-25. Grounds M., Partridge T.A., Sloper J.C. The contribution of exogenous cells to regenerating skeletal muscle: an isoenzyme shidy of muscle allografts i n mice. J. Pathology 1980; 132: 325-341. Watt, D.J., Morgan, J.E., Partridge, T.A. Allografts of muscle precursor cells persist in the non-tolerized host. Neuromuscular disorders 1991; 1: 345-355. 28. Roy, R., Dansereau, G., Tremblay, JS.Belles-Ides, , M., Labrecque, C. and Bouchard, J.P. Major histocompatibility complex (MHC) antigens expression on human myoblasts. Transplantation Proceedings 1991; 23: 799-801. 29. Grounds, M.D., Lai, MC., Fan, Y-, Codling, J.C. and Beilharz, M.W. Transplantation in the mouse model - the use of a Y-chromosomespecific DNA clone to identify donor cells in situ. Transplantation 1991; 52: 1101-1105. 30. Fan, Y., Maley, M., Belharz, M. and Grounds, M. Rapid death of injected myoblasts in myoblast tramfer therapy. Muscle and nerve (in press). 31. Fan, Y., Beilharz, M., Grounds, LM.A potential alternative strategy for myoblast transfer therapy: The use of sliced muscle grafts. Ce11 Transplantation 1996; 5: 421-429. Fluorescence intensity MYOBLAST TRANSPLANTATlON RESULTS, CELLULAR AND HUMORAL REACTION OF DIFFERENT üROUt>S Ub ALLOGRAFT AND SYNGENEIC GRAET Group Legend: Donor suain MAC C57BU10J 1 weak male transpl 3 5 we ak cells per fiel R cl0 + 10-50 ++ 50-100 +++ >IO0 nos R ++ + + + + ++ + ++ / number of miir 1. Bilan des essais diniques effectués chez l'humain À ce jour, cinq projets cliniques majeurs concernant la transplantation myoblastes ont été menés en Amérique du Nord et en Italie afin de traiter garçons atteints de dystrophie musculaire de Duchenne. La décision procéder aux essais cliniques a été prise par les cliniciens lors d e la réunion sur la thérapie de transfert de myoblastes organisée par l'Association Américaine pour la Dystrophie musculaire qui a eu lieu à New York en 1989. La plupart des chercheurs présents à cette réunion se sont farouchement opposés à cette décision argumentant que les essais chez l'humain étaient prématurés et que trop peu d'études chez l'animal avaient été faites. Néanmoins, la gravité des symptômes de la DMD d'une part et l'ignorance quant à l'immunogénicité des cellules myoblastiques d'autre part ont incité certaines équipes à tenter des transplantations chez l'humain. Cette situation a provoqué de grandes controverses mais les premières conclusions suite à ces expériences sont: I'échec à remplacer efficacement la dystrophine dans les muscles atteints et qu'aucune augmentation de la force musculaire n'a été démontrée. Malgré ces insuccès, aucun dommage sérieux n'a été remarqué chez les garçons dystrophiques suite aux greffes. Toutefois, il est important de spécifier que des études chez la souris ont démontré des dommages «bystander» au muscle hôte (Wernig, 1995) c'est-à-dire que la réponse immunitaire envers le greffon peut simultanément endommager les tissus environnants. Par ailleurs, le problème avec les essais cliniques c'est le peu d'informations disponibles concernant le destin des myoblastes injectés. Il est possible de prélever une biopsie musculaire au site d'injection certes, mais seule une petite portion du muscle greffé peut ainsi être évaluée. Il est éthiquement et tediniquement impossible de mieux suivre l'évolution de la greffe dans les muscles dystrophiques déjà sévèrement compromis. 2. Retour à l'expérimentation chez l'animal Le destin des myoblastes injectés peut heureusement être étudié chez les modèles animaux. Il existe deux modèles importants pour la DMD: le chien et la souris. La plupart des expérimentations sont effectuées sur cette dernière et démontrent un certain succès. Par ailleurs, ces transplantations sont souvent effectuées dans des conditions très différentes de celles entourant les greffes chez l'humain. En effet, l'étude de plus de trente articles s u ce sujet a révélé que la majorité des transplantations a été faite sur des animaux hôtes «traités» (Grounds, 1996). Dans treize de ces études, les myoblastes ont été injectés dans des souris nude ou SCID dont le système immunitaire n'est pas fonctionnel pour le rejet des cellules étrangères. Dans neuf des études, les muscles hôtes ont été pré-irradiés permettant ainsi aux myoblastes importés de mieux participer à la régénération. L'irradiation au site de la greffe semble favoriser grandement la prolifération cellulaire et même le déplacement des myoblastes injectés. Onze des articles concernaient l'injection de lignées cellulaires telle C2C12 au lieu des myoblastes. Malheureusement ces cellules, en fusionnant avec les fibres musculaires hôtes, ont démontré qu'elles pouvaient former des tumeurs in vivo. Ces expérimentations faites dans des conditions «optimales» sont intéressantes mais peu applicables à la situation clinique. De plus, le succès de ces études n'expliquent pas l'inçuccès des essais thérapeutiques. Il devient nécessaire d'étudier les transplantations de myoblastes dans des conditions qui se rapprochent le plus de celles rencontrées lors des greffes chez l'humain. C'est ce que nous avons tenté en étudiant le l'implication des antigènes mineurs d'histocompatibilité lors de transplantations de myoblastes entre individus histocompatibles. En effet, le système immunitaire demeure une barrière difficilement surmontable et les AgMH ont une grande part de responsabilité dans le rejet immunitaire. 3. Importance des AgMH Les antigènes mineurs d'histocompatibilité ont d'abord été identifiés par leur capacité à induire des rejets de tumeurs et de transplantations de tissu. La présente étude démontre que les AgMH peuvent également être responsables d u rejet des myoblastes lors de transplantations chez la souris mdx. Antérieurement, le groupe du Dr. Tremblay a démontré qu'il est possible d'obtenir un très bon succès de greffe lors de transphntations syngéniques. Par ailleurs, lorsque des transplantations sont effectuées entre donneur et receveur différents au niveau d'un ou de plusieurs antigènes d'histocompatibilité, on obsewe seulement quelques fibres exprimant de la dystrophine, 4 semaines après la greffe. Ces fibres positives ont habituellement disparu vers la seizième semaine suivant la greffe- La présence de rares fibres à ce moment peut être due à des mutations inverses plutôt qu'aux transplantations elles-mêmes. Parmi ces même groupes de souris transplantées, on a pu observer par immunohistochimie une infiltration cellulaire importante composée de cellules immunitaires (lymphocytes CD4+, CD8+, macrophages, neutrophiles). Selon les combinaisons de transplantations, les différences au niveau de la quantité de fibres positives pour la dystrophine et du degré de l'infiltration cellulaire peuvent s'expliquer par un rejet plus ou moins rapide variant selon l'irnmunogénicité des antigènes impliqués. Quoi qu'il en soit, la présence de cellules immunitaires à 4 et à 16 semaines suivant les greffes sont l'évidence d'un rejet et on peut conclure à un rejet chronique contrairement à la réponse envers les antigènes du CMH qui est beaucoup plus rapide et qui s'apparente plutôt au rejet aigu. Par ailleurs, l'activation des cellules d'infiltration a été confirmée par la détection des ARNm de l'interféron gamma (EN-y) et de la granzyme B. Ces protéines ne sont exprimées que par les cellules activées. Suite aux transplantations allogéniques nous avons détecté des anticorps dirigés contre les myoblastes greffés. Étonnamment, les transplantations syngéniques semblent également avoir induit la formation d'anticorps. C'est d'autant plus surprenant que les antigènes mineurs sont caractéris& par leur incapacité à favoriser la formation d'anticorps. Certains auteurs d o ~ e n un t sens plus large au terme AgMH en les définissant comme des peptides provenant de la protéolyse de n'importe quelle protéine, en autant qu'ils soient capables de lier une molécule de CMH et de provoquer le rejet du tissu transplanté. On peut ainsi désigner la dystrophine comme un AgMH. Lors des transplantations syngéniques, seule cette protéine absente chez l'hôte aurait pu induire la formation d'anticorps. Toutefois, cette hypothèse ne peut être retenue car la dystrophine, n'étant exprimée que par un faible pourcentage de myoblastes matures, ne peut être impliquée dans la réaction du sérum de l'hôte avec les myoblastes du donneur utilisés pour le test de cytofluorimétrie. De plus, la dystrophine étant une protéine inhacellulaire, elle demeure nondisponible pour les anticorps lors du contact avec des cellules nonperméabilisées. L'absence de réaction entre les sera des receveurs et les myoblastes des donneurs cultivées en absence de semm de veau fœtal (FCS) nous permet de croire que les anticorps pourraient être dirigés contre des composés retrouvés dans le milieu de culture. Ces composés se retrouveraient à la surface des cellules cultivées en présence de FCS et provoqueraient la formation d'anticorps au moment de la greffe. Nous avons confirmé cette interprétation par ELJSA. En effet, les sera provenant de souris transplantées 4 semaines plus tôt ont fourni des réactions positives avec le FCS contrairement aux sera des souris naïves qui se sont avérées toutes négatives pour la présence d'anticorps anti-FCS. I1 est possible que ces anticorps soient à l'origine de la positivité des réactions croisées entre les sera post-greffes et les myoblastes cultivés en présence de FCS utilisés pour les tests de cytofluorimétrie. Par ailleurs, la plupart des transplantations allogéniques ont donné des réactions positives pour la présence d'anticorps contre les cellules greffées. Certains de ces anticorps pourraient être dirigés contre des antigènes mineurs autres que la dystrophine. En effet, même si ces antigènes sont habituellement définis en terme de médiation cellulaire, il reste possible que certains puissent induire une réaction humorale. Une autre hypothèse qui reste cependant à vérifier est que les myoblastes, lors de leur passage en culture et au contact avec le FCS, pourraient se mettre à exprimer des antigènes absents dans d'autres conditions. Finalement, il est important de souligner que ces anticorps anti-FCS ne semblent pas perturber le succès des greffes puisque leur présence dans les greffes syngéniques n'a pas causé de rejet. La réaction immunitaire cellulaire dirigée contre les myoblastes obtenus à partir de donneurs incompatibles pour un OU pour plusieurs antigènes mineurs a été clairement démontrée par l'expression des ARNm de I'IFN-y et de la granzyme B par les cellules immunitaires d'infiltration. Cette réaction est probablement dirigée contre des AgMH. 11 est à remarquer que les transplantations syngéniques sont exemptes de ce type de réaction. L'utilisation d'agents immunosuppresseurs devient donc nécessaire lors de transplantations de myoblastes et ce, même lorsque le receveur est histocompatible avec le donneur. Les AgMH sont présentés aux lymphocytes en association avec les molécules du CMH de classe 1. Il a déjà été démontré que les myoblastes expriment ces molécules (Roy et al., 1995). Ceci pourraient expliquer pourquoi les myoblastes sont rejetés relativement rapidement lors des transplantations entre donneurs et receveurs compatibles uniquement pour le CMH. Le rejet des transplantations de myoblastes possédant l e chromosome Y effectuées chez des femelles démontrent que la différence au niveau de quelques antigènes mineurs (ceux reliés au chromosome Y) est suffisante pour mener au rejet immunologique. 4. Autres études se rapportant au rejet causé par les AgMH D'autres équipes ont également entreprit des études de transplantation de cellules musculaires s'apparentant à mon projet. Il est difficile de comparer les résdtah car les expériences ont été menées de diverses façons. De plus, ces équipes n'avait pas établit d'avance l'étude du rôle des AgMH dans le rejet de greffe. C'est suite aux mauvais résultats obtenus qu'ils ont imputé la responsabilité du rejet immunologique aux AgMH. Le groupe de Fan ( Muscle and nerve, sous presse) ont obtenu d'excellents résultats (survie du greffon: 1 ans et plus) lors de transplantations syngéniques en greffant des muscles entiers provenant de souris mâles et greffés dans des femelles. Mentionnons que le but des transplantations dans des femelles était de suivre I'évolution de la greffe en se sentant d'une sonde spécifique au chromosome Y. Ceci contraste avec les pauvres résultats qu'ils ont obtenus lors de transplantations de myoblastes isolés. L'inaccessibilité du muscle entier au système immunitaire, du moins pour la réponse immunitaire immédiate, pourrait expliquer ces résultats. Des expériences similaires avaient été effectuées par Grounds en 1980. Les résultats obtenus contrastent avec ceux du groupe précédent puisqu'aucune greffe n'a sunrécu plus de 7 jours. Toutefois, puisqu'ils ne se sont pas préoccupés du sexe des receveurs, les antigènes mineurs reliés au chromosome Y (H-Y) ont pu être impliqués dans le rejet. Le degré d'immunogénicité et la quantité des antigènes présents pourraient donc expliquer ces divergences. Enfin, le groupe de Watt a obtenu des résultats fort surprenants. En effet, dans les allotransplantations de précurseurs myoblastiques, ils ont observé que 86% des transplantations avaient un temps de s u ~ allant e jusqu'à 100 jours selon les combinaisons de souches utilisées qui sont d'ailleurs différentes des nôtres. Encore une fois, on peut expliquer ces divergences par le degré d'immunogénicité des antigènes en présence (Watt, 1991). 6. Perspectives d'avenir Comme le démontre la présente étude, le système immunitaire demeure un obstacle majeur pour la réussite des transplantations de myoblastes, même lorsque le donneur et le receveur sont histocompatibles. L'utilisation d'agents immunosuppresseurs (FK 506, CyA) demeure nécessaire. Malheureusement, ces composés entraînent des effets secondaires indésirables. Ils n'agissent pas de façon spécifique, ils requièrent une administration à long terme et augmentent les risques d'infection. La forme idéale d'immunosuppression thérapeutique devrait être administrée sur une courte période et rendre l'individu tolérant aux antigènes désirés à long terme sans toutefois empêcher la réponse à un agent infectieux. L'équipe du Dr. Tremblay travaille présentement sur l'utilisation des anticorps monoclonaux (mAb) comme agents immunosuppresseurs. Il existe de nombreuses études concernant l'emploi de mAb lors de transplantations chez différents modèles animaux (Cobbold et al., 1986; Madsen et al., 1987; Lenschow et al., 1995). La plupart des mAb utilisés ont été choisis selon leur interaction avec les récepteurs de membrane impliqués dans la CO-stimulation et l'adhésion des cellules immunitaires tels: CD3, CD4, CDS, ICAM-1, UA-1, B7-1 et 87-2. Le bloquage de ces récepteurs sur les CFA ou sur les lymphocytes T par les mAb adéquats peut mener soit au rejet immunitaire, soit à l'immunosuppression ou encore à la tolérance à long terme (Waldmann et al., 1989; Pearson et al., 1990; Bushel et al., 1995). Cette dernière situation est celle qui serait idéale et que l'on cherche à obtenir. Il reste à déterminer si cette forme d'immunosuppression serait efficace dans le cas d'incompatibilité au niveau des AgMH. Cette voie de recherche semble très prometteuse pour contrer le système de surveillance immunitaire et nous permet d'espérer pouvoir un jour effectuer des transplantations de myoblastes dans les musdes des patients dystrophiques et ainsi remplacer la protéine absente. Arahata K, Ishiura S, Ishiguro T, Tsukahara T, Suhara Y, Eguchi CH, Ishihara, Nonaka 1, Ozawa E, Sugita H. Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne muscle dystrophy peptide. Nature 1988; 333: 861-63. Bailey DW. Genetic histocompatibility in mice. Immunogenetics 1975; 2: 249-56. Bailey DW. How pure are inbred strains of mice? Immun01 Today 1982; 3: 310. Bjorkman PJ, Parham P. Structure, function and diversity of dass-1 major histocompatibility complex molecules. A m . Rev. Biochem 1990;59: 253-88. Blau HM, Webster C, Chiu CP, Guttrnan S, Chandler F. Differenciation properties of pure populations of human dystrophie muscle cells. Exp Cell Res 1983;144:495503. Boris LK, Temin HM. Recent advances in retrovirus vector technology. Curr. Opin. genet. Dev. 1993;3: 102-9. Boyd Y, Munro E, Ray P. et al. Molecular heterogenecity of handocations associated with muscular dystrophy. Clinical Genetics 1987; 31: 265-272- Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature 1989;338:259-62. Chelley J, Kaplan JC, Marie P,Gautron S, Kahn A. Transcription of the dystrophin gene in human muscle and non-muscle tissue. Nature 1988;3335358 -860. Clemens P,Caskey T. Gene therapy prospects for Duchenne Muçcular Dystrophy. Eur. Neurol1994; 34: 181-5. Davies KE,Pearson PL, Harper PS. et al. Linkage analysis of two cloned DNA sequences flanking the Duchenne muscular dystrophy locus on the short arrn of the human X-chromosome. Nuclei Acids Research 1983;11:2303-2312. Desnuelle C. Les enseignements de la dystrophine. Le Presse médicale 1994; 19:896-900. Dudienne GB. Recherches sur la paralysie musculaire pseudohypertrophique ou paralysie myosdérosée. Ar& Gen Med 1868;11,179,305,421,552. Dunckley MG, Love DR, Davies KE, Walsh FS, Morris GE, Didcson G. Retroviral-mediated transfer of a dystrophin mînigene into mdx m o u e myoblasts in vitro. FEBS Lett. 1992;296: 128-134. England SB, Nicholson LV, Johnson MA et al. Very mild muscular dystrophy assoaated with the deletion of 46% of dystrophin. Nature 1990; 343: 180-2. Falk K, Rotzsdike O, Deres K et al. Identification of naturally processed viral nonapeptides allows their quantification in infected cells and suggests an allele-specific T celi epitope forecast. J Exp Med 1991;174:425-34. Gordon RD, Simpson E, Samelson LE. In vitro celi mediated immune response to the male specific antigen in mouse. J Exp Med 1975; 142:1108-20. Gorer PA, Lyman S, Snell GD. Studies on the genetic and antigenic basis of tumor transplantation. Linkage betweeen a histocompatibility gene and b e d in mice. Proc. R. Soc. Lond (Biol.) 1948;135: 499-505. Goulmy E, Termijtelen A, Bradley BA, Van Rood JJ. Alloimmunity to hurnan H-Y. Lancet 1976;2: 1206. Goulmy E, Termijtelen A, Bradley BA, Van Rood JJ. Y-antigen killing by Tcells of women is resticted by HLA. Nature 1977; 266: 544-5. Goy JJ,Stauffer JC, Deruaz JE'et al. Myopathy as possible side-effect of cyclosporin. Lancet 1989; 1: 1446-7. Hohlfeld R, Engel AG. Induction of HLA-DR expression on human myoblasts with interferon-gamma. Am J Pathol. 1990; 136: 503-8. Huard J, Labrecque C, Dansereau G, et al. Dystrophin expression in myotubes formed by the fusion of normal and dystrophic myoblasts. Muscle and nerve 1991; 14: 178-82. Horowitz M. Adenoviridae and their replication. In virology. Edited by Fields BN, Knipe DM. New York: Raven press; 1990: 1679-1740. Law PK,Goodwin TG, Wang MG. Normal myoblast injections provide genetic treatment for murine dystrophy. Muscle and nerve 1988; 11:52533. Grounds M. Commentary on the present state of knowledge for myoblast transfer therapy . CeU transpkmtation 1996; 3: 431-3. Guérette Br Roy R, Tremblay M, Asselin 1, Kinoshita 1, Puymirat J, Tremblay J. Increased granzyme B mRNA after ailoincompatible myoblast transplantation. Transplantation 1995; 60: 1011-6. Guérette Br Vilquin JT, Guigras M, Wood K, Tremblay J. Prevention of immune reaction triggered by first-generation adenoviral vectors by monoclonal antibodies and CTLA4Ig. Human gene therapy 1996; 7: 1455-63. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: The protein product of ; the Dudieme muscular dystrophy locus. Cell 1987; 51: 91: 928. Hoffman EP, Fishbeck KH, Rosenberg P, Pollina C, Kunkel LM. Cell and fiber-type distribution of dystrophin. Neuron 1988; 1:411-20. Hoffman EP. Human molecular genetics and the elucidation of the primary biochemical defect in Duchenne muscular dystrophy. Ce11 Motil. Cytoskel 1989; 14:163-8. Johnson LL. At how many histocompatibility loci do congenic mouse strains differ. Probability estimates and some implications. J Hered 1987 2: 27. Kaminsky E, Sharrock C, Hows J. Frequency analysis of CTL precursor: Possible relevance to HU-matched unrelated donor bone marrows transplantation. Bone Marrow Transplantation 1988; 3: 149-65. h o T. FK 506, a novel immunosuppressant isolated from a streptomyces. J Antibiot. 1987; 40: 1249-55. Kinoshita 1, Vilquin JT,Guérette B, Asselin 1, Roy R, Tremblay JP. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle and nerve 1994; 17: 1407-15. Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod shape cytoskeletal protein. Ce11 1988; 53: 219-26. Komgold R, Wettstein PJ. Immunodominance i n the GVHD T cell response to minor histocompatibility antigens. J of Imrnunol. 1990; 145: 4079-88. Kuby J. Immunobiology. W.H. Freeman and Company 1992;230-39. Kunkel LM, Monaco AP, Middlesworth W, Ochs HD, Lah SA. Speafic doning of DMD fragments absent from the DNA of a male patient with a X-chromosome deletion . Proc. Natl. Acad. Sci. U.S.A. 1985;82: 4778-82. Labrecque C, Bouchard JP,Malouin F, Roy R, Huard J, Tremblay JP. Approches thérapeutiques de la myopathie de Duchenne par transplantation de myoblastes. Méd S u 1991;7:821-9. Law PK,Goodwin TG, Li HJ. Histoincompatible myoblast injection irnproves muscle structure and function of dystrophie mice. Tramp1 Proc. 1988;20: 1114-19. Loveland B, Simpson E. The non-MHC transplantation antigens: Neither weak nor minor. Immun01 Today 1986;7: 223. Lyon MF. Gene action in the X-diromosorne of the mouse (Mus.musculus L.) Nature 1961;190:372-4. Mohanakumar T. The role of MHC and non-MHC antigens in alIograft immunity . Medical Intelligence Unit 1994;l-104. Monaco AP, Neve RL, Coletti-Freener, Bertlson CJ, Kumit DM, Kunkel LM. Isolation of candidate cDNAs for portion of the Duchenne muscular dystrophy gene. Nature 1986;323: 646-50. Morgenstern JI?, Land H,. Advance mamrnalian gene transfer : High titre retroviral vectors with multiple dmg selection markers and a complementary helper free packaging cell lines. Nucleic Acids Res. 1990;19: 3587-96. Morgan JE. Normal muscle precursor implants restitute normal histology to degenerating muscles of the mdx mouse. J Neurol Sci. 1990;98: 126. Mulligan RC. The basic saence of gene therapy. Science 1993;260: 926932. Murray JM, Davies KE,Harper PS.et al. Linkage relationship of a cloned DNA sequence on the short a m of the X-chromosome to Duchenne muscular dysfxophy. Nature 1982;300:69-71. Pagel CN, Morgan JJ5Myoblast Transfer and gene therapy in muscular dystrophies. Micros. Res. and Tech. 1995;30:469-79. Partridge TA, Morgan JE,Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophic-negative to positive by injection of normal myoblasts. Nature 1989; 337:176-9. - Partridge TA, Sloper JC. A host conhibution to the regeneration of muscle grafts. J Neurol Sci 1977;33: 425-35. Partridge TA. Invited review: myoblast transfer: a possible therapy for inherited myopathies? Muscle and nerve 1991;14: 197-212. Perreault C, Decary G, Brochu S et al. Minor histocompatibility antigens. Blood 1990;76(7): 1269. Rammensee HG, Falk K, Rôtzschke O. MHC molecules as peptide receptors. Curent Opinion in Immunology 1993; 5: 35-44. Rammensee HG, Falk K, Rotzschke O. Peptides naturally presented by MHC class 1molecuIes. Ann. Rev. Immunol. 1993; 11: 213-244. Rammensee HG, Klein J. Polymorphism of minor histocompatibility genes in wild rnice. Immunogenetics 1983; 17: 637. Roopenian DC. What are minor histocompatibility loci? A new look at a old question. Immun01 Today 1986; 7: 223. Roy R, Tremblay J, Huard J, Richards C, Malouin F, Bouchard JP. Antibody formation after myoblast hansplantation in Duchenne dystrophie patients, donor HLA-compatible. Scornik JC, Brunson ME, Howard RJ et al. AUoimmunization, memory and the interpretation of cross-matdi results for rend transplantation. Transplantation 1992;54: 389-94. Sincinski P, Geng Y, Ryder-Cook AS et al . The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 1989; 244: 1578-80. Simpson E, Chandler P, Goulmy E et al. Çeparation of the genetic loci for the H-Y antigen and the testis determination on the human Ychromosome. Nature 1987; 326: 876-8. Simpson E, Tomonan K, Lovering E. Minor transplantation antigens: Their role in shaping the T cell repertoire. Immun01 Lett 1989; 21: 39. Simpson E. T and B lymphocytes: Two repertoires or one? Immun01 Lett 1986;12: 185. Snell GD, Dausset J, Nathenson S. Histocornpatibility. San Diego, CA, Academic 1976. Snell GD. Methods for the study of histocompatibility genes. J Genetics 1948; 49: 87-108. Snell GD. Studies in histocompatibility. Science 1981; 213: 172-8. Soulillou J.P. Les greffes d'organes. La recherche 1986; 177: 586-99. Stephew FE, Tyler FH. Studies in disorders of muscle. The inheritance of diildhood progressive muscular dystrophy in 33 kindreds. Am J of Hum Gen 1951; 111-125. Stredni B, Schwartz RH. Alloreactivity of an antigen specific T-tell clone. Nature 1980; 287 855-7. Townsend A, Elliot T, Cenindolo V et al. Assembly of MHC class 1 molecules analyzed in vitro. Cell 1990; 62: 285-95. Townsend A, Ohlen Cf Bastin J, Ljunggren HG, Foster L, Karre K. Association of class 1major histocompatibility heavy and light chains induced by viral peptides. Nature 1989; 340: 443. Tremblay JP,Bouchard JI?, Malouin F et al. Myoblast transplantation between monozygotic twin girl carriers of Duchenne muscular dystrophy. Neuromuscular Disord 1993; 3: 583-92. Tremblay JF, Malouin F, Roy R et al. Results of a triple blind clinical study of myoblast transplantations wi thout immunosuppressive treabnent in young boys with Duchenne muscular dystrophy. Cell Transplantation 1993; 2: 99-112. Vilquin JT,Asselin 1, Guérette B et al. Myoblast allotransplantation in mice: the degree of success varies depending on the efficacy of vanous irnrnunosuppressive treatments. Transpl. Proceed. 1994; 6: 337273. Viquin JT, Kinoshita 1, Roy R, Trembiay JP. Cydophosphamide immunosuppression does not permit successful myoblast allotransplantation in moue. Neuro. disord. 1995; 5: 511-517. Vilquin JT,Asselin 1, Guérette 8,et al. Successful myoblast allotransplantation in mdx mice uskg rapamycin. Transplantation 1995; 59: 422-426. Vilquin JT,Wagner E, Kinoshita I f Roy R, Trernblay JP. Successful histocompatible myoblast transplantation in dystrophin-deficient mdx m o u e despite the production of antibodies against dystrophin. J Cell Bi01 1995; 131: 975-88. Von Boehmer H, Haas W. H-2 restricted cytolytic and non-cytolytic T cell clones: isolation, specificity and functional analysis. Immun01 Rev 1981; 54: 27-56. Waldmann H, Cobbold S. The use of monoclonal antibodies to adueve immunological tolerence. TiPS 1993; 14: 143-48. Wallny HJ, Rammensee HG- Identification of classical minor histocompatibility antigens as cell derived peptide. Nature 1990; 343: 275-81Nattrass FJ.On the classification, natural history and treatment Walton JN, of the myopathies. Brain 1954; 77: 169-231. Watkins DC,Hoffman EP, Slayter HS, Kunkel LM. Immunoelectron rnicroscopic localization of dystrophin in myofibres. Nature 1988; 333: 863-6. Watt DJ, Morgan JE, Clifford MA, Partridge TA. The movement of muscle precursor cells between adjacent regeneration muscles in the mouse. Anat Embryol 1987; 175: 527-36. Wernig A, Irintchev A. Bystander damage of host muscie caused by implantation of MHC-compatible myogenic cells. J Neurol Sci. 1995; 130: 190-6. Wettstein PJ, Bailey DW. Immunodominance in the immune respowe to multiple histocompatibility antigens. Immunogenetics 1982; 16: 47-58. Wettstein PJ. Immunodominance in T cell responce to multiple non-H2 histocompatibility antigens II: Observation of hierarchy among dominant antigens. Immunogenetics 1986; 24: 24. Wettstein PJ. Minor histocornpatibility loci. Litwin S. ed. Human immunogenetics. New York: Dekker 1989; 339-57. Yewdell JVV, Bennidc JR. The b i n q logic of antigen processing and presentation to T ce&. Cell 1990;62:2036. Zubrzycka-Gaam EE, Bulman DE, Karpati G, et al. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 1988; 333: 466-9. IMAGE EVALUATION TEST TARGET (QA-3) APPLIED IMAGE. lnc 1653 East Main Street -.- Rochester. NY 14609 USA -- ---, Phone: 71648.2-0300 Fax: 716/28&5989