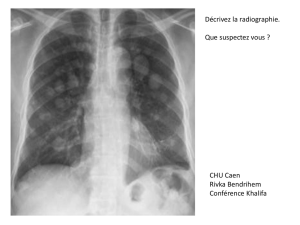
Adénocarcinome ductal pancréatique et micro

Bases fondamentales
Adénocarcinome ductal
pancréatique et micro-
environnement tumoral :
rôle de la réaction
desmoplasique fibreuse
Corinne Bousquet, Christiane Susini
Inserm U858, Institut de Biologie Moléculaire de Rangueil, IFR31, CHU Rangueil,
Toulouse, France
Bien que longtemps ignoré dans la régulation de la progression
tumorale, le rôle promoteur du micro-environnement tumoral est
désormais reconnu par de nombreuses études. Ce micro-
environnement appelé aussi stroma-tumoral est principalement
composé de tissus connectifs fibreux, la matrice extracellulaire,
ainsi que d’une composante cellulaire représentée par des fibro-
blastes, des cellules inflammatoires et immunitaires et des cellules
composant les vaisseaux sanguins. Il existe une communication
étroite entre cellules épithéliales cancéreuses et cellules stromales.
Les cellules cancéreuses sont en effet capables d’altérer leur micro-
environnement de manière à le rendre plus propice au développe-
ment tumoral en favorisant la mise en place d’un stroma « réactif »,
appelé également réaction desmoplasique, correspondant à une
production excessive de tissu connectif fibreux et à une activation
de chacune des composantes cellulaires du stroma, avec principa-
lement apparition de myofibroblastes, mais également de réactions
inflammatoires, immunitaires et angiogéniques. Cette réaction stro-
male contribuerait en contrepartie à la progression tumorale. Parmi
les tumeurs malignes caractérisées par une forte réaction stromale,
l’adénocarcinome ductal pancréatique, un cancer très agressif de
très mauvais pronostic, présente comme signature histopathologi-
que une infiltration fibreuse abondante retrouvée aussi bien dans la
tumeur primaire que dans les métastases lymphatiques et hépati-
ques. Cette revue a pour objectifs de caractériser les mécanismes
moléculaires et cellulaires responsables de la mise en place de la
réaction desmoplasique dans l’adénocarcinome ductal pancréati-
que en insistant exclusivement sur la composante fibreuse de cette
réaction, d’établir son rôle dans la progression de ce type de
cancer, et d’évaluer le bénéfice thérapeutique potentiel qu’appor-
terait le ciblage du micro-environnement tumoral pour le traitement
de ce type de cancer.
Hépato-Gastro, vol. 14, n°3, mai-juin 2007
Tirés à part : C. Bousquet
231
doi: 10.1684/hpg.2007.0101
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

Il est actuellement reconnu que le micro-environnement tumoral joue un rôle critique sur la progression
tumorale, ce qui est particulièrement vrai pour l’adénocarcinome ductal pancréatique qui présente une forte
réaction desmoplasique fibreuse. Cette réaction provient d’une communication étroite et réciproque entre
cellules épithéliales cancéreuses et cellules du stroma, les cellules épithéliales altérant leur micro-
environnement de manière à le rendre plus propice à leur propre prolifération, survie, migration et invasion.
Mots clés : adénocarcinome ductal pancréatique, réaction desmoplasique fibreuse, micro-environnement
Notre appréciation restreinte de la progres-
sion tumorale en tant qu’un enchaînement
d’événements rythmé par l’accumulation de
mutations dans les cellules épithéliales cancéreuses a
largement contribué à l’ignorance du micro-
environnement comme promoteur du phénotype malin.
Toutefois, depuis deux décennies ont commencé à se
multiplier les études démontrant la participation du
micro-environnement dans la progression tumorale. En
effet, la tumeur est désormais reconnue comme tissu
hétérogène et complexe de part sa composition en
types cellulaires différents correspondant à la fois aux
cellules épithéliales cancéreuses per se et aux cellules
du tissu environnant appelé stroma tumoral. Ce stroma
correspond à une structure de tissu connectif composée
d’une matrice extracellulaire (MEC), la matrice tumo-
rale, ainsi que d’une composante cellulaire correspon-
dant à des fibroblastes, des cellules inflammatoires et
immunitaires (lymphocytes, macrophages et mastocy-
tes), et des cellules composant les vaisseaux sanguins
(cellules endothéliales, péricytes et cellules musculaires
lisses). Il existe une communication étroite entre cellules
épithéliales cancéreuses et cellules stromales, chaque
composante régulant la dynamique de l’autre affectant
ainsi la croissance tumorale. Il a été démontré que les
cellules cancéreuses sont capables d’altérer leur micro-
environnement de manière à le rendre plus propice et
favorable au développement tumoral. La résultante est
la mise en place d’un stroma « réactif », appelé égale-
ment réaction desmoplasique, correspondant à une
production excessive de tissu connectif fibreux et à une
activation de chacune des composantes cellulaires du
stroma, avec principalement apparition de myofibro-
blates ou CAF (carcinoma-associated fibroblasts), mais
également de réactions inflammatoires, immunitaires et
angiogéniques. Cette réaction stromale contribuerait
en contrepartie à la progression tumorale. En effet, au
cours des états cancéreux, une perturbation des
contacts des cellules cancéreuses entre elles et des
cellules cancéreuses–MEC favorise la migration et
l’invasion tumorale.
D’autre part, la MEC, dont la composition est modifiée,
sert de réservoir aux facteurs de croissance et aux
cytokines sécrétés par les cellules cancéreuses et/ou
les cellules stromales, favorisant ainsi leur interaction
avec leur récepteur respectif, et régulant leur activation
et/ou turnover [1].
L’importance relative du stroma tumoral varie considé-
rablement d’un type tumoral à un autre, mais n’est pas
a fortiori corrélée au degré de malignité de la tumeur
[1]. Parmi les tumeurs malignes caractérisées par une
forte réaction stromale, l’adénocarcinome ductal pan-
créatique présente comme signature histopathologique
une infiltration fibreuse abondante retrouvée aussi bien
dans la tumeur primaire que dans les métastases lym-
phatiques et hépatiques. Il s’agit d’un cancer extrême-
ment agressif caractérisé par une progression très
rapide et l’apparition précoce de métastases, et de
pronostic très sévère en raison du diagnostic qui se fait
dans la plupart des cas à un stade avancé d’évolution
de la maladie et de la faible réponse des cellules
cancéreuses pancréatiques à la chimiothérapie ou à la
radiothérapie [2].
Cette revue a pour objectifs de caractériser les mécanis-
mes moléculaires et cellulaires responsables de la mise
en place de la réaction desmoplasique dans l’adénocar-
cinome ductal pancréatique en insistant exclusivement
sur la composante fibreuse de cette réaction, d’établir
son rôle dans la progression de ce type de cancer, et
d’évaluer le bénéfice thérapeutique potentiel qu’appor-
terait le ciblage du micro-environnement tumoral pour le
traitement de ce type de cancer.
Caractéristiques du stroma tumoral
fibrotique de l’adénocarcinome
ductal pancréatique
La MEC présente dans l’adénocarcinome ductal pan-
créatique est principalement composée de collagènes,
de glycoprotéines non collagéniques, d’élastine et de
protéoglycannes. La réaction fibreuse desmoplasique
observée dans ce type de cancer se traduit, d’une part,
par une augmentation de la production de la MEC,
notamment de fibronectine, de protéoglycannes mais
surtout de collagène interstitiel fibrillaire (de type I et III)
dont l’expression est augmentée d’environ 3 fois par
rapport à ce qui est observé dans le pancréas sain, et
d’autre part par une perte de la membrane basale,
exposant ainsi les cellules cancéreuses pancréatiques
Bases fondamentales
Hépato-Gastro, vol. 14, n°3, mai-juin 2007
232
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

directement au collagène interstitiel, ce qui entraîne
une perturbation des interactions cellules épithéliales-
–MEC [3]. Les collagènes fibrillaires contribuent nor-
malement au maintien de la structure des organes
épithéliaux en apportant une force de tension à la
matrice interstitielle, elle-même séparée des cellules
épithéliales par la membrane basale, une matrice
extracellulaire spécialisée. Cette réaction desmoplasi-
que est associée à une multiplication intense des cellu-
les stromales fibroblastiques, qui dans certains cas de
cancer sont même en surnombre par rapport aux
cellules épithéliales cancéreuses elles-mêmes. Ces cel-
lules d’origine mésenchymateuse appelées cellules stel-
laires pancréatiques (PSC) correspondent à des fibro-
blastes différenciés en un phénotype de
myofibroblastes activés ou CAF [4]. En immunohisto-
chimie, les cellules fibroblastiques sont spécifiquement
reconnaissables par des immunomarquages dirigés
contre les protéines desmine ou protéines de liaison de
l’acide rétinoïque, et contre la protéine actine amus-
culaire lisse (aSMA), une fois que ces cellules sont
activées en myofibroblastes. Une analyse comparative
de la MEC présente dans des lésions de pancréatite
chronique et d’adénocarcinome ductal pancréatique a
montré une forte homologie structurale dans leur com-
position, impliquant une source cellulaire commune de
production. Le rôle crucial des PSC a d’ailleurs été
clairement démontré dans la pathogénie de la pan-
créatite chronique au cours de laquelle une accumula-
tion de collagène fibrillaire participe au développe-
ment de la maladie [5]. Par des comarquages en
immunohistochimie avec des anticorps aSMA, colla-
gène type I, III et fibronectine, et/ou en hybridation in
situ avec une sonde reconnaissant l’ARNm du procol-
lagène alpha1I, ces myofibroblastes ont été identifiés
comme étant la source principale de production du
collagène et de fibronectine présents dans la réaction
desmoplasique pancréatique [6, 7]. La preuve en est
que les tumeurs issues de la xénogreffe de cellules
cancéreuses pancréatiques chez l’animal ne présen-
tent en l’absence de PSC qu’une très faible réaction
fibreuse [7]. Des PSC isolées à partir d’échantillons
humains d’adénocarcinome pancréatique ont été
immortalisées, et leur utilisation facilitera l’étude des
interactions cellules cancéreuses–PSC dans l’avenir [8]
.
Facteurs sécrétés par les cellules
cancéreuses pancréatiques
influençant la réaction
desmoplasique fibreuse
Facteurs de croissance
Il a été bien démontré que les cellules cancéreuses
pancréatiques surexpriment de nombreux facteurs de
croissance et leurs récepteurs, et sécrètent ces facteurs
qui, par autocrinie, vont promouvoir leur prolifération.
D’autre part, de plus en plus nombreuses sont les
études démontrant le rôle stimulant des cellules cancé-
reuses épithéliales pancréatiques sur la prolifération et
la synthèse de matrice extracellulaire par les PSC
(figure 1). De cette manière, la tumeur détourne le
micro-environnement hôte local à son avantage pour
favoriser sa propre croissance et sa survie. Des expé-
riences de culture de cellules fibroblastiques soit en
présence des milieux conditionnés, soit directement en
coculture avec les cellules cancéreuses pancréatiques
ont en effet montré la sécrétion, par les cellules cancé-
reuses épithéliales, de facteurs de croissance qui ainsi
stimulent par paracrinie les PSC. De la même manière,
l’activation des PSC observée au cours des états de
pancréatite chronique et responsable de la réaction
stromale observée dans cette pathologie, résulterait
d’une stimulation par des facteurs de croissance sécré-
tés par les cellules acineuses injuriées, voire les pla-
quettes ou les macrophages activés [7]. Certains fac-
teurs fibrogéniques surexprimés et sécrétés par les
cellules cancéreuses pancréatiques ont été identifiés et
seraient responsables des effets inducteurs sur la proli-
fération et la synthèse de MEC par les PSC. Il s’agit
notamment du PDGF (platelet-derived growth factor)et
du FGF-2 (fibroblast growth factor) qui accélèrent la
prolifération des PSC, et qui avec le TGFb(tumor
growth factor) sont également responsables de l’effet
de synthèse de MEC [7, 9]. En plus de ces effets positifs
sur la prolifération et la synthèse de MEC des PSC, le
FGF-2 est un facteur pro-angiogénique en revanche
son effet stimulant sur la croissance des cellules endo-
théliales [10]. Le TGFb, une cytokine sécrétée dans le
milieu extracellulaire sous forme latente non active,
peut être activé par liaison avec l’intégrine avb6,
présente à la surface des cellules cancéreuses pancréa-
tiques [11]. Il a été démontré que le TGFb, dont
l’expression est augmentée dans les cancers pancréa-
tiques et est corrélée à un stade tumoral avancé et à un
mauvais pronostic, stimule non seulement la production
de MEC par les cellules fibroblastiques, mais modifie la
composition de cette matrice en favorisant l’expression
de versicane, un protéoglycanne à effet pro-invasif au
détriment des protéoglycannes à effet antitumoral
décorine et du lumicane dont l’expression est diminuée
par cette cytokine [3]. De plus, le TGFbréduit ou
augmente respectivement le niveau d’expression des
métalloprotéinases matricielles (MMP) ou de leurs inhi-
biteurs (TIMP, tissue inhibitor of MMP), avec comme
résultante une inhibition de la dégradation de la MEC
et un effet global pro-fibrotique. D’autre part, le TGFb
stimule l’activation des cellules fibroblastiques pan-
créatiques en myofibroblastes exprimant la protéine
a-SMA ce qui est associé à une forte augmentation de
Hépato-Gastro, vol. 14, n°3, mai-juin 2007 233
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

l’expression du récepteur PDGF, favorisant ainsi la
boucle de stimulation paracrine cellule cancéreu-
se–PSC dépendante du PDGF [7, 9]. Des travaux ont
d’autre part montré que la transfection de l’ADNc du
TGFbdans les cellules cancéreuses pancréatiques
PANC-1, où cette cytokine est peu exprimée de
manière endogène, entraîne une forte réaction desmo-
plasique après greffe de ces cellules dans le pancréas
de souris nude, non observée après greffe des cellules
contrôles non transfectées, et associée à une surexpres-
sion de collagène de type I et de fibronectine [9]. Enfin,
il a été démontré une forte corrélation entre l’expres-
sion du TGFbet celle du collagène de type I dans les
tumeurs humaines d’adénocarcinome pancréatique, et
la signalisation de cette cytokine a été trouvée augmen-
tée dans les grades 3 de ces tumeurs, ainsi illustrés par
une augmentation de phosphorylation de Smad2, une
protéine directement activée par liaison au complexe
TGFbrécepteur TbRI/TbRII [12]. L’ensemble de ces
résultats démontrent donc une forte implication du
TGFbdans la mise en place de la réaction stromale
fibreuse dans l’adénocarcinome ductal pancréatique.
Cette réponse fibroblastique serait attribuable à des
effets directs mais également indirects du TGFbqui
stimule notamment l’expression du connective tissue
growth factor (CTGF), une glycoprotéine sécrétée de la
famille CCN (Cyr61, CYsteine-Rich 61 ; CTGF ; NOV
(nephroblastoma overexpressed gene). Effecteur de
l’effet fibrotique du TGFb, le CTGF est un puissant
mitogène des fibroblastes et des cellules musculaires
lisses [9, 13]. Une analyse comparative de l’expres-
sion génique de cellules stromales et épithéliales can-
céreuses dans des sites primaires de cancer pancréa-
tique a montré une expression différentielle de certains
gènes seulement exprimés dans les cellules stromales
ou épithéliales cancéreuses pancréatiques, dont le
CTGF spécifiquement exprimé au niveau épithélial
[14]. Une fois sécrété par les cellules épithéliales
cancéreuses pancréatiques sous l’action du TGFbpar
exemple, le CTGF participe donc à la mise en place de
la réaction stromale fibroblastique en activant les PSC
via le récepteur intégrine a5b1 [15].
Métalloprotéases matricielles
En plus des facteurs de croissance, les cellules épithé-
liales cancéreuses sécrètent des MMP, enzymes protéo-
lytiques qui vont permettre un remodelage de la MEC,
et notamment une destruction de la membrane basale,
favorisant ainsi la migration et l’invasion locale puis à
distance par les cellules cancéreuses. La dégradation
de molécules matricielles par ces enzymes expose
certains domaines cryptiques protéiques générant
ainsi de nouveaux fragments moléculaires pouvant
présenter des fonctions pro- ou anti-migratoires et
angiogéniques. Les MMP activent également des fac-
teurs de croissance en favorisant leur libération de la
membrane cellulaire et/ou de la MEC où ils étaient
jusque-là séquestrés sous forme latente [16] (figure 1).
Les collagènes interstitiels, incluant le type I, principaux
composants de la MEC de l’adénocarcinome ductal
pancréatique, sont fortement résistants à la protéolyse,
de part leur structure en triple hélice et leur organisa-
tion fibrillaire. Par des études génétiques utilisant des
souris délétées pour le gène codant pour la
métalloprotéinase-1 de type membranaire (MT1-
MMP), il a été démontré que la MT1-MMP est le
principal régulateur de la collagénolyse interstitielle,
ces souris présentant des troubles sévères de crois-
sance résultant de leur incapacité à dégrader les
collagènes interstitiels durant la formation des os et
tissus mous [17]. D’autre part, des études d’expression
génique dans les cellules stromales et cancéreuses
pancréatiques présentes dans les sites d’invasion pri-
maires ou métastatiques d’adénocarcinome pancréati-
que ont montré que MT1-MMP est la principale colla-
génase interstitielle surexprimée dans les cellules
épithéliales cancéreuses, et que son expression est
augmentée dans les lésions métastatiques par rapport
aux lésions primaires d’adénocarcinomes pancréati-
ques, et ce plus particulièrement dans des régions où la
réaction fibreuse est importante [14].
Les PSC influencent les cellules
cancéreuses pancréatiques
Expériences de coculture cellules
épithéliales cancéreuses–PSC
Une fois activé par les cellules cancéreuses, il semble-
rait que le stroma tumoral, en retour, envoie des
signaux oncogéniques vers les cellules cancéreuses en
stimulant la progression tumorale. Mises en présence
de myofibroblastes tumoraux « activés », des cellules
épithéliales cancéreuses pancréatiques acquièrent des
propriétés invasives accrues [18], voire développent
une résistance exacerbée à une certain nombre de
drogues chimiothérapeutiques [19] ; ce résultat est
également observé lorsque des cellules épithéliales
pancréatiques non cancéreuses mais immortalisées
sont cultivées en présence de PSC, ces cellules épithé-
liales étant capables de se transformer et d’acquérir un
phénotype malin [12]. Ce phénomène est généralisa-
ble à d’autres tumeurs épithéliales, notamment les
carcinomes prostatiques ou squameux cellulaires de la
peau [1, 20, 21]. D’autre part, des expériences utili-
sant des fibroblastes irradiés de manière à engendrer
des dommages sublétaux à l’ADN, ont conforté le rôle
de ces cellules dans la progression tumorale, puisque
leur coculture avec des cellules cancéreuses pancréati-
ques augmente le pouvoir invasif des cellules cancéreu-
Bases fondamentales
Hépato-Gastro, vol. 14, n°3, mai-juin 2007
234
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

ses [22]. Ces résultats suggèrent donc que la survenue
de cancers après irradiation serait la résultante de
mutations apparues non seulement dans les cellules
épithéliales mais également dans les fibroblastes du
stroma. Ces résultats sont également en accord avec
l’observation que des fibroblastes humains en sénes-
cence sont capables de transformer des cellules épithé-
liales en culture et de promouvoir la formation de
tumeurs chez l’animal [23]. L’acquisition de mutations
dans ces cellules avec l’âge leur apporterait un phéno-
type comparable à celui observé dans les myofibro-
blastes tumoraux, avec notamment l’acquisition de
propriétés de sécrétion d’un certain nombre de facteurs
favorisant la progression tumorale.
Facteurs sécrétés par les PSC influençant
les cellules épithéliales cancéreuses
Les myofibroblastes tumoraux favorisent la progression
tumorale de différentes manières (figure 1). Tout
d’abord, ils sécrètent des composants de la MEC à
activité pro-migratoire et/ou invasive. Les protéines de
la MEC sont en effet fortement impliquées dans la
régulation de nombreux processus cellulaires tels que
la survie, la migration, l’invasion ou la différenciation ;
la preuve en est qu’en l’absence de MEC, les cellules
épithéliales adhérentes entrent en apoptose. Cette inter-
action entre MEC et cellules épithéliales repose sur des
interactions dynamiques entre des protéines de la MEC
Induction de la réaction desmoplasique fibreuse :
• transformation des fibroblastes en myofibroblastes (PSC)
• prolifération des PSC (sécrétion PDGF)
Acquisition d’un phénotype malin des cellules épithéliales :
survie, migration, invasion, chimiorésistance
Cellule épithéliale
cancéreuse pancréatique
Myofibroblaste pancréatique
(PSC)
Facteurs de croissance
et leurs récepteurs
Récepteur intégrine
MEC
MMP
TIMP
Mobilisation de facteurs de croissance
et de molécules biologiquement actives de la MEC
par les MMP
Figure 1.Interactions entre cellules épithéliales et cellules stromales fibroblastiques entretenant la réaction desmoplasique et le phénotype
cancéreux malin.Les cellules épithéliales cancéreuses sécrètent des facteurs de croissance et des protéases agissant par autocrinie et paracrinie
sur les cellules fibroblastiques du stroma. En conséquence, d’une part, les cellules épithéliales prolifèrent davantage ce qui entretient et amplifie
le phénomène de sécrétion de ces facteurs, et d’autre part, les cellules fibroblastiques sont différenciées en myofibroblastes ou PSC (pancreatic
stellate cells) et présentent une prolifération accrue. Ces PSC sécrètent une grande quantité de protéines de la MEC dont la composition est
modifiée, ce qui conduit au développement d’une forte réaction desmoplasique fibreuse fortement observée dans l’adénocarcinome
pancréatique. Les PSC sécrètent également des facteurs de croissance qui vont favoriser leur propre prolifération mais également la survie,
migration, invasion et chimiorésistance des cellules épithéliales cancéreuses. D’autre part, les MMP sécrétées dans le milieu extracellulaire à
la fois par les PSC et par les cellules épithéliales cancéreuses vont dégrader la MEC, en favorisant la migration et l’invasion cellulaire,
mobilisant des facteurs de croissance séquestrés dans cette matrice, et générant l’apparition de nouvelles molécules biologiquement actives à
effet pro- ou anti-migratoire et angiogénique.
Hépato-Gastro, vol. 14, n°3, mai-juin 2007 235
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.
 6
7
8
6
7
8
1
/
8
100%