TP évalué - Accueil / steber.svt

Doc 1 : La mucoviscidose
En France, un nouveau-né sur 4200 est touché par la mucoviscidose. Environ 200 enfants
naissent chaque année en France avec la mucoviscidose.
Grâce aux projets de la recherche et de soins, pour les enfants qui naissent en 2008,
l’espérance de vie est de 46 ans, alors qu'elle n'était que de 7 ans en 1965. Mais l’âge moyen
de décès de l’ensemble des patients n'est que de 28 ans.
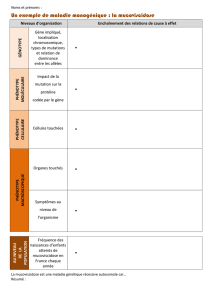
La mucoviscidose est une maladie génétique qui touche les voies respiratoires et le système
digestif. Elle n’est pas contagieuse.
La maladie peut s’exprimer de façon différente chez chaque patient. Certains sont plus
touchés au niveau des poumons et d’autres au niveau de l’appareil digestif.
Ce mot est composé de : MUCUS + VISCOSITÉ = MUCOVISCIDOSE.
Le corps de chacun d’entre nous produit du mucus. Cette substance fluide tapisse et humidifie
les canaux de certains organes de notre corps. Dans le cas de la mucoviscidose, le mucus est
épais et collant. Ce manque de fluidité va provoquer des difficultés au niveau des voies
respiratoires et digestives.
Lorsque les poumons sont sains, le mucus qui tapisse les parois des voies respiratoires est
fluide et veille à la protection de celles-ci et des alvéoles. Chez une personne atteinte de
mucoviscidose, le mucus est épais et visqueux. Il adhère aux parois des bronches, gênant
ainsi le passage de l’air. Si le mucus reste dans les bronches, il favorise la croissance d’agents
infectieux (virus et surtout bactéries) provoquant ainsi des infections.
Les voies et canaux digestifs (intestins, pancréas, foie) peuvent également être obstrués,
provoquant des problèmes de digestion.
http://www.vaincrelamuco.org/
Doc 2 : le test de la sueur
Il consiste à mesurer le taux de chlorures dans la sueur. Les résultats sont interprétés
de la manière suivante :
Taux de chlorures
Résultat du
test
inférieur à 40 mmol.L-1
négatif
compris entre 40 mmol.L-1 et 60
mmol.L-1
douteux
supérieur à 60 mmol.L-1
positif

Doc 3 : la protéine CFTR
http://www.stanford.edu/class/psych121/humangenome-CF.htm
Doc 4 : coupes transversales de bronches chez un individu sain et un individu
malade (bordas 280)
SVT 1S, Bordas
Mucoviscidose
Dans la partie « Variation génétique et santé » du programme, il est indiqué qu’il s’agit de montrer
aux élèves que la détermination des causes d’une maladie n’est possible qu’en utilisant un mode
de pensée statistique tout en ne considérant que les principes généraux d’une approche
épidémiologique.
L’étude des phénotypes liés à la lactase permet de dégager ces principes avec notamment les
notions de risque relatif et de taux de concordance chez les jumeaux.
On peut appliquer ces principes à l’approche des maladies inscrites au programme, mucoviscidose
et maladies multifactorielles (diabète, maladies cardiovasculaires et autres). Dans cet aperçu, on
se limite à la mise en évidence de l’implication de facteurs génétiques, mais une démarche du
même type peut être utilisée pour les facteurs d’environnement.

La mucoviscidose
Aujourd’hui, le fait que la mucoviscidose soit une maladie héréditaire, et plus précisément une
maladie monogénique due aux mutations du gène CFTR situé sur le chromosome 7, ne fait pas de
doute. Mais il n’en a pas toujours été ainsi.
La mucoviscidose a été définie à la fin des années 1930 par Dorothy Andersen qui a réuni dans sa
diagnose les manifestations respiratoires et pancréatiques. Trois hypothèses ont été émises en ce
qui concerne l’origine de la maladie : un déficit nutritionnel, notamment en vitamine A durant la
grossesse ou la jeune enfance, une origine infectieuse, une origine héréditaire. Cette dernière
hypothèse repose sur le constat de la présence de plusieurs enfants atteints de mucoviscidose
dans quelques familles. Mais cela pouvait aussi s’expliquer par les autres hypothèses. En 1946,
Andersen et Hodges publient les résultats d’une étude épidémiologique qui validait l’hypothèse
génétique.
Des données épidémiologiques : mise en évidence d’une origine génétique
Les chercheurs sont partis des enfants qui sont venus en consultation à l’hôpital des enfants
malades de New York entre 1938 et 1945 et chez lesquels on a diagnostiqué une mucoviscidose. A
partir de là, ils ont fait une étude familiale pour chacun des enfants atteints en déterminant
exactement l’état des sœurs et frères de ces enfants : atteints de mucoviscidose ou pas. On dit
que l’enfant malade est le proposant à partir duquel est conduite l’étude familiale. Les familles
avec un seul enfant (l’enfant malade), non informatives, n’ont pas été prises en compte.
Sur 31 familles ainsi sélectionnées, il y a eu en tout 58 frères et sœurs des enfants malades, et
parmi ceux-ci 13 atteints de mucoviscidose. Bien entendu dans toutes ces familles, les parents
n’étaient pas malades.
Pour exploiter ces données, on peut utiliser le calcul du risque relatif qui est le rapport de la
prévalence chez les apparentés (ici frères et sœurs) des proposants sur la prévalence de la
mucoviscidose dans la population générale.
A l’époque, dans l’état de New-York, la prévalence de la mucoviscidose à la naissance était de
1/2500. La prévalence de la mucoviscidose chez les apparentés des proposants est de 13/58.
Le risque relatif est donc de 13/58 / 1/2500 soit environ 560.
Un risque relatif aussi élevé indique une maladie à forte composante génétique.
La mucoviscidose : maladie monogénique
Il s’agit ensuite de trouver le modèle génétique qui rend compte des données. Le modèle
génétique le plus simple est celui où la maladie est due aux allèles morbides d’un seul gène. On se
place dans le cadre de ce modèle. Puisque les parents des enfants malades ne sont pas atteints
cela signifie qu’il s’agit d’une maladie récessive, donc que les allèles à l’origine de la mucoviscidose
sont récessifs. Les élèves doivent alors pouvoir indiquer les génotypes des parents et préciser que
la descendance de nombreux couples d’hétérozygotes doit comprendre 25% d’enfants atteints de
mucoviscidose. Les données de l’étude précédemment citées indiquent 13 enfants atteints sur 58
soit 22,4% ce qui est proche de 25%. Le modèle monogénique est validé par cette étude familiale.
Génotypes d’une famille
Le gène en cause a été identifié et séquencé en 1989. Situé sur le chromosome 7, il a été appelé
CFTR en raison du rôle joué par la protéine qu’il code (Cystic Fibrosis Transmembrane
conductance Regulator).
Avec les informations et vos savoirs faires, proposez une argumentation détaillée en
faveur d'une origine génétique de la maladie.
(Un fichier numérique me sera envoyer à la fin de la séance)

Fichiers disponibles sur le site Chap6 :
CFTR_sauvage.pdb
CFTR_mutante.pdb
Famille-CFTR.edi
1
/
4
100%