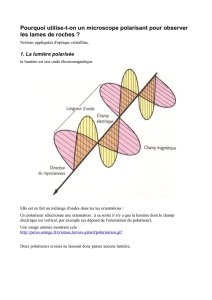
Etude expérimentale de la cristallisation du

Université Libre de Bruxelles (ULB)
Faculté des Sciences Appliquées / Ecole Polytechnique
Service de Chimie Industrielle
THESE
Présentée en vue de l’obtention du titre de
DOCTEUR EN SCIENCES APPLIQUEES
par :
Yi ZHU
Etude expérimentale de la cristallisation du
bicarbonate de sodium
Promoteur de thèse :
Prof. Marie-Paule DELPLANCKE - OGLETREE
Examinateurs :
M. J. Amouroux Professeur (ENSCP, Paris VI)
M. Th. Cartage Ingénieur (Solvay – Bruxelles)
M. J-L. Delplancke Professeur (ULB – Bruxelles)
Mme M-P. Delplancke – Ogletree Professeur (ULB – Bruxelles)
Mme V. Halloin Professeur (ULB – Bruxelles)
Mme C. Porte Professeur (CNAM – Paris)
M. S. Van Vaerenbergh Chargé de cours (ULB –Bruxelles)

La passion du progrès

Remerciements
Ces recherches ont été effectuées au laboratoire du Service de Chimie
Industrielle de la Faculté des Sciences Appliquées de l’Université Libre de Bruxelles
(ULB), dirigé par Madame le Professeur M-P. Delplancke-Ogletree. Je tiens à lui
exprimer ma reconnaissance pour l’intérêt qu’elle a porté à ce travail pour m’avoir
permis de suivre une formation scientifique de qualité et pour ses encouragements.
Cette thèse n’aurait pu être possible sans le financement de la société Solvay.
Ici se trouve l’expression de mes remerciements les plus sincères.
J’exprime toute ma gratitude à Monsieur P. Demilie de la société Solvay s.a.
Je le remercie pour ses conseils scientifiques, pour sa sympathie et pour les 700 kg de
bicarbonate de sodium qu’il nous a apporté pendant ces quatre années de recherche.
Cette thèse est le fruit d’une collaboration entre la société Solvay s.a. et
l’Université Libre de Bruxelles. Je tiens à exprimer mes remerciements à Madame P.
Davoine, à Monsieur T. Cartage et à Monsieur F. Coustry de la société Solvay s.a.,
pour leur confiance, leurs encouragements et l’intérêt constant qu’ils ont porté à ce
travail.
Ma profonde gratitude s’adresse également à Monsieur C. Breton, à Monsieur
C. Criado, à Monsieur L. Ninane, ainsi qu’à toute l’équipe du CER Solvay Dombasle
pour m’avoir apporté leurs connaissances et expériences. Je voudrais remercier
spécialement Mademoiselle N. Hirschauer et Monsieur E. Roy pour leurs aides
essentielles au début de ma thèse.
De sincères remerciements à Monsieur le Professeur P.H. Duvigneaud et à
Monsieur le Professeur G. Schmitz du Service de Chimie Industrielle, pour leurs
disponibilités, leurs conseils scientifiques et leur sympathie.
Je pense à tous mes amis du Service de Chimie Industrielle, spécialement à
Monsieur L. Szabo, Mademoiselle T. Segato, Monsieur P. Madau, Monsieur A. Diard,
Monsieur C. Aughuet, Monsieur Y. Ducenne, Madame D. Van Der Schrick et
Mademoiselle M. El Mkhtrioui, pour leurs soutiens techniques indispensables. Je tiens
aussi à remercier Madame M. Neuckermans, Mademoiselle N. Vercruysse et Madame
C. de Scoville pour les services administratifs rendus. Je remercie Mademoiselle E.
Nguyen pour les corrections d’orthographe de cette thèse. Merci à Monsieur G. Van
Rompaey, Monsieur C. Vanoerbeek, Monsieur G. Wallaert ainsi que Madame C-G.
Jung pour leurs aides amicales.
Je tiens également à remercier Monsieur J. De Maeyer, souffleur de verre à
l’ULB, pour son travail de qualité et ses aides techniques indispensables.
Je remercie toute l’équipe du Service de Génie Chimique, dirigé par Madame
le Professeur V. Halloin, spécialement Monsieur H. Baudine pour son aide technique,
Monsieur B. Haut pour les échanges développés au cours de nos thèses, Monsieur H.
Saddaoui pour son aide amicale et Madame C. Hanon pour sa sympathie.
Je tiens à remercier toute l’équipe du Service de Génie Métallurgique, dirigé
par Monsieur le Professeur L. Segers, spécialement Monsieur M. D’Hoker, Monsieur

A. Schepens, Monsieur R. Defour, Monsieur F. Pucci pour leurs aides techniques et
leur gentillesse.
J’exprime ma gratitude à Monsieur le Professeur J-L Delplancke et son équipe
du Service de Science des Matériaux et Electrochimie, spécialement à Monsieur J.
Dille pour ses aides sur l’utilisation de l’analyse d’images et à Monsieur L. Canet
pour sa sympathie.
De sincères remerciements à Monsieur S. Van Vaerenbergh du service de
Chimie Physique pour avoir accepté d’être membre du comité d’accompagnement de
ma thèse et pour l’attention qu’il a portée à cette thèse.
Que Monsieur H.J.M. Kramer, Professeur associé du Laboratory of Process
Equipment de Delft University of Technology et Monsieur P. Bowen du Laboratoire
de Technologie des Poudres de l’Ecole Polytechnique Fédérale de Lausanne (EPFL)
soient remerciés pour leurs conseils scientifiques et leur sympathie.
Je voudrais remercier Monsieur H. Brequel (Vesuvius) pour la discussion sur
la cristallographie.
Je voudrais également remercier Monsieur J. Amouroux, Professeur de
l’Ecole Nationale Supérieure de Chimie Paris (ENSCP), Monsieur A. Delacroix,
Professeur du Conservatoire National des Arts et Métiers (CNAM), Mademoiselle C.
Porte, Professeur de l’Université Pierre & Marie Curie (CNAM), Monsieur A.
Rannou, Directeur technique de la société ARD-SOLIANCE, Monsieur M. Moscosa-
Santillan, Maître de Conférence à l’ENSCP et Madame B. Loï Mi Lung – Somarriba
du CNAM pour m’avoir conduit au domaine de la cristallisation industrielle, pour les
formations qu’ils m’ont apportées pendant mes études à Paris et pour leurs amitiés.
Merci à mes ex-collègues belges du laboratoire comme Monsieur S. Poellaer
(Seghers), Monsieur O. Conreur (UCB Chimie), Monsieur N. Havelange (Henogen),
Monsieur X. Dallenogare (Ecologiste), Mademoiselle N. Alaoui (Euroscreen),
Monsieur B. De.Bontridder (Lhoist), Monsieur O. Barbery (Holcim) et Mademoiselle
C. Masselus (Province du Hainaut), pour les grandes amitiés belgo-chinoises
développées au cours de ces quatre années.
Je remercie tous particulièrement Monsieur O. Van Volden pour son amitié.
Mes remerciements et mes amitiés s’adressent à Madame L. Vanderkerken-
Wierzchucka, ainsi que ses amis. Je la remercie pour m’avoir permis de loger dans sa
maison, tout près de ULB, pendant ces quatre années de thèse, pour les attentions et
les conditions qui m’ont été offertes.
Enfin, je ne pourrai pas oublier ma famille. Je la remercie pour ses
compréhensions et ses encouragements.

Y. ZHU (11-2004) i
Table des matières
Introduction générale 1
Chapitre I. Notions fondamentales de la cristallisation du bicarbonate de sodium
I-1. Solution 8
I-1-1. Solubilité, sursaturation et zone métastable 8
I-1-1-1. Solubilité 8
I-1-1-2. Sursaturation 10
I-1-1-3. Zone métastable : le diagramme solubilité - sursaturation 12
I-1-2. Solution de bicarbonate de sodium 13
I-1-2-1. Ions en solution aqueuse 13
I-1-2-2. Equilibre du bicarbonate de sodium dans l’eau 14
I-1-2-3. Courbe de solubilité du bicarbonate de sodium 16
I-1-2-4. Caractérisation des propriétés d’une solution du bicarbonate
de sodium 17
I-2. Cristaux de bicarbonate de sodium 17
I-3. Germination 18
I-3-1. Germination primaire homogène 19
I-3-2. Germination primaire hétérogène 23
I-3-3. Germination secondaire 25
I-3-4. Expression de la cinétique de germination 27
I-4. Croissance 27
I-4-1. Mécanismes de croissance 27
I-4-2. Croissance en solution aqueuse 29
I-4-3. Morphologie des cristaux en solution aqueuse 30
I-5. Agglomération et bris de grains 31
1-5-1. Agglomération 31
1-5-1-1. Mécanismes d'agglomération 31
1-5-1-2. Cinétique d’agglomération 32
1-5-1-3. Agglomération dans la pratique 34
1-5-2. Bris de grains ou attrition 35
Chapitre II. Application de la densimétrie aux études de la cristallisation du
bicarbonate de sodium
II-1. Détermination de la concentration en bicarbonate de sodium hors ligne 36
II-1-1. Méthode potentiométrique 36
II-1-2. Méthode volumétrique 37
II-1-3. Utilisation d’un indicateur 37
II-1-4. Méthode thermo-gravimétrique 38
II-2. Techniques d’étude de la cristallisation en continu 38
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
1
/
257
100%