En détail ici

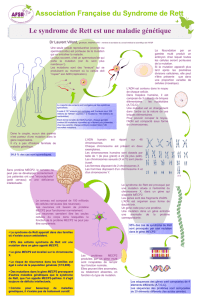
Comment traiter le syndrome de Rett ?
L-Carnitine, importante pour le métabolisme osseux et l’énergie
cellulaire. Essais cliniques en 1999, 2001 et 2005. Pas d’effet majeur
observé.
Mélatonine, importante pour la régulation du rythme veille / sommeil.
Essai clinique en 1998 pendant 4 semaines avec une amélioration du
sommeil chez certaines patientes.
Naltrexone, inhibiteur de certains récepteurs dans le cerveau.
Essai clinique en 1994 avec des effets secondaires négatifs.
Tyrosine et tryptophane, acides aminés nécessaires pour la
synthèse de certains messagers chimiques dans le cerveau.
Essai clinique en 1990 sans effet observé.
Bromocriptine, stimulant de certains récepteurs dans le cerveau.
Essai clinique en 1990, pas d’effet majeur observé.
PHARMACOLOGIE
Remplacer le gène MECP2 déficient.
Des études pré-cliniques indiquent que la réintroduction d’un gène
MECP2 normal chez la souris modèle (malade) grâce à la
manipulation des embryons peut faire disparaître les signes cliniques
de l’animal.
Cette technologie n’est pas applicable à la thérapie génique humaine.
Plusieurs équipes (USA / France) tentent donc d’utiliser des vecteurs
viraux pour délivrer un gène MECP2 normal dans les cellules
malades.
La difficulté est d’atteindre le cerveau et de traiter suffisamment de
cellules pour espérer obtenir un effet thérapeutique.
Les résultats chez l’animal ne sont pas attendus avant plusieurs années.
Remplacer la protéine MeCP2 déficiente.
Plutôt que de remplacer le gène, certaines équipes (Autriche /
Allemagne) tentent de fabriquer de la protéine MeCP2 et de la
délivrer dans le cerveau.
Des études cellulaires préliminaires sont en cours. Les études chez l’animal
sont en cours et une application aux malades n’est pas envisageable avant
plusieurs années.
Dans ces deux cas, un problème majeur pour le traitement est que
l’organisme traité développe une réponse immunitaire contre le
traitement (virus ou protéines étrangères).
THÉRAPIE GÉNIQUE & PROTÉIQUE
DEMAIN ?
P
Inhibiteurs d’histones déacétylases (HDAC), molécules qui peuvent
modifier la structure de l’ADN pour conserver certains gènes sous forme
active.
Leur spécificité et leur toxicité sont mal évaluées et leurs effets
secondaires ne sont pas connus.
Des études sur des cellules de malades (in vitro) sont en cours.
Agents déméthylants, molécules qui vont modifier la structure de l’ADN
pour empêcher la répression de certains gènes. Il n’existe pas d’agents
spécifiques de certains gènes, leur effet est global. Ils sont utilisés pour
traiter certains cancers.
Des études sur des cellules de malades (in vitro) sont en cours.
Modificateurs de mutation, molécules qui peuvent « supprimer » l’effet
de certaines mutations (mutations non-sens). Leur toxicité à long terme
n’est pas connue. Des études sur des cellules de malades (in vitro) sont
en cours.
Des essais cliniques ont débuté pour d’autres pathologies (ex :
myopathie de Duchenne) mais ont été arrêtés.
PHARMACOLOGIE
Vitamine B9, importante pour le métabolisme des acides aminés.
Essai clinique en 2009 avec un effet modeste sur l’épilepsie de 3
filles (sur 12 traitées).
Fluoxétine, modulatrice de la synthèse de certains messagers
chimiques (sérotonine) dans le cerveau.
Essai clinique en 2010, interrompu pour cause d’effets secondaires
mineurs.
Désipramine, modulatrice de la synthèse de certains messagers
chimiques (noradrénaline) dans le cerveau.
Essais pré-cliniques positifs chez l’animal modèle.
Essai clinique en cours en 2011, résultats non disponibles.
IGF-1, facteur de croissance.
Essais pré-cliniques positifs chez l’animal modèle.
Essai clinique prévu pour 2012.
Il existe deux grandes catégories de traitement que l'on peut envisager pour le syndrome de Rett.
• La première catégorie concerne les médicaments qui vont agir sur les conséquences de la maladie : troubles respiratoires, troubles du sommeil,
agitation, épilepsie... Ces médicaments ne sont pas spécifiques au syndrome de Rett.
• La seconde catégorie concerne les traitements qui vont agir directement sur la cause de la maladie : remplacement du gène ou de la protéine
dans les cellules malades. Ils seront spécifiques au syndrome de Rett.
A l'heure actuelle, seules les approches pharmacologiques ont été testées dans le syndrome de Rett, hélas sans beaucoup de succès.
De nouveaux traitements sont à l'essai (soit dans les modèles animaux de la maladie, soit chez des patients dans le cadre des essais cliniques) en
utilisant des molécules connues ou nouvelles. Pour les molécules nouvelles, l'application clinique ne sera possible qu'à long terme (il faudra tester au
préalable la toxicité et la tolérance, l'apparition d'effets secondaires ou non désirés). Pour les molécules connues (déjà utilisées dans des
médicaments), l'application clinique chez les patients pourrait être plus rapide.
Les approches de thérapie génique ou protéique, quant à elles, sont en cours de mise au point chez l’animal de laboratoire. Elles ne pourront pas
être proposées aux malades avant plusieurs années.
HIER & AUJOURD'HUI
AFSR version avril 2011
1
/
1
100%