DCEM1 Génétique Médicale

1
Faculté de Médecine Purpan
¯¯¯¯¯¯¯¯¯¯¯¯
Année universitaire 2008-2009
DCEM1
Génétique Médicale
E. Bieth,
G. Bourrouillou,
N. Chassaing,
P. Calvas

2
Plan
1°) Génétique clinique
1. Abord du malade en génétique, modes de transmissions et histoires familiales,
2. Examen clinique et notions de dysmorphologie et de syndromologie génétique
2°) Cytogénétique
1. Les anomalies chromosomiques, épidémiologie et signes d’appels,
2. Les examens chromosomiques, leurs indications, leur interprétation
3°) Génétique Moléculaire
1. Le diagnostic moléculaire, ses indications,
2. Les stratégies et l’interprétation,
3. Les règles et les modalités de prescription
4°) Diagnostic prénatal
1. Indications, modalité, limites

3
1 Génétique clinique
1.1 Abord du malade en Génétique clinique
1.1.1 Préambule
La génétique médicale est la discipline qui s’intéresse aux maladies génétiques notamment
dans leur dimension familiale. Il faut rappeler néanmoins que si toutes les maladies
héréditaires ont un déterminisme génétique en revanche la majorité des maladies génétiques
ne sont pas héritées (ex : cancers par mutations somatiques, maladies orphelines par néo-
mutations,…). La mise en évidence du caractère transmissible d’une affection tient une place
essentielle en médecine prédictive et préventive. Elle influe, parfois de façon déterminante,
sur la prise en charge médicale des patients et de leur famille et permet à travers le conseil
génétique des choix reproductifs éclairés. C’est pourquoi, il convient dès lors que l’on
suspecte une maladie génétique de rechercher systématiquement dans les antécédents
familiaux s’il existe ou non des éléments évocateurs d’une prédisposition héréditaire. Cette
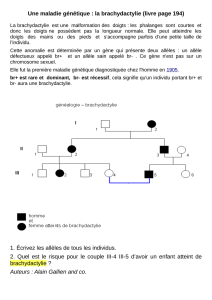
recherche consiste en pratique à dresser à l’aide des symboles connus un arbre généalogique
précis (figure 1). Le sujet atteint pour qui un diagnostic a été initialement établi est appelé cas
index ou propositus, il est logiquement le point de départ de l’enquête généalogique. Ce
diagnostic de maladie génétique ou supposée telle repose classiquement sur des critères
phénotypiques et/ou génotypique ou caryotypique. On entend par phénotype l’ensemble des
caractères observables c’est-à-dire des éléments, pathologiques ou non, recueillis par
l’approche clinique. Cette dernière inclue classiquement la recherche des symptômes
fonctionnels grâce à l’interrogatoire, des symptômes dits objectifs grâce à l’examen physique
complet et enfin des signes seulement détectables par des examens paracliniques (imagerie
médicale, explorations en électrophysiologie, en biochimie…). L’approche clinique en
génétique médicale n’est donc pas fondamentalement très différente de celle des autres
spécialités mais, elle s’appuie à chacune des trois étapes de la démarche diagnostique, sur des
points particuliers qui lui confèrent une certaine spécificité. L’importance des antécédents
familiaux recueillis par l’interrogatoire a déjà été soulignée. Lors de l’examen physique on
s’attachera à rechercher plus particulièrement des anomalies morphologiques même minimes
qui peuvent constituer de précieux éléments d’orientation. Ainsi, les données de l’enquête
généalogique d’une part et la description de critères dysmorphiques (dysmorphologie) d’autre
part contribuent à restreindre le champ des hypothèses diagnostiques et à orienter la
prescription des examens paracliniques. Ces derniers visent essentiellement en génétique
médicale à étudier les caractéristiques génétiques : caryotype grâce aux analyses de
cytogénétique et génotype grâce aux analyses de génétique moléculaire. Il convient de
souligner qu’en dehors du caryotype, il n’existe pas aujourd’hui d’examen permettant
d’appréhender de façon globale les caractéristiques génétiques d’une personne. La
prescription de ces examens ne peut donc être systématique mais raisonnée c’est-à-dire guidée
par l’ensemble des éléments sémiologiques recueillis par un examen clinique minutieux. Or,
la sémiologie en génétique médicale est, nous allons le voir, riche, variée et complexe…
1.1.2 Spécificités de l’interrogatoire, enquête familiale et évaluation du caractère
transmissible.
1.1.2.1 Les données de l’interrogatoire sont cruciales tant pour la recherche d’une étiologie
génétique que pour l’évaluation du risque de récurrence d’une affection au diagnostic parfois
incertain et pour laquelle se pose la question du caractère transmissible ou pas. C’est une

4
étape de l’approche clinique aussi essentielle que celle de l’examen physique. L’interrogatoire
en génétique médicale doit, tout en étant concis, viser à l’exhaustivité : des renseignements a
priori d’importance mineure peuvent avoir dans certaines affections syndromiques complexes
une valeur capitale pour l’orientation diagnostique. Il doit aussi être adapté au consultant dont
l’émotivité, les barrières psychologiques (tabous familiaux par exemple) et la capacité de
compréhension (barrière linguistique par exemple) sont autant de difficultés à communiquer.
Enfin, l’interrogatoire doit être orienté : en fonction de l’affection concernée il conviendra de
bien faire préciser les circonstances qui ont conduit à sa découverte ainsi que son évolution
dans le temps (maladie fixée ou évolutive…) et de rechercher dans l’histoire familiale des
éléments susceptibles d’être en rapport avec la maladie.
La situation la plus emblématique est celle du jeune enfant présentant un retard des
acquisitions associé ou non à des anomalies malformatives. Il conviendra ici d’être
particulièrement vigilant aux antécédents maternels ainsi qu’aux circonstances de la grossesse
et de la période périnatale. Certaines données orientent en effet vers des causes acquises
responsables de phénocopies (on entend par phénocopies, des affections au phénotype
comparable mais d’origine -acquise ou innée- différente). La prise de toxiques pendant la
grossesse (alcool, médicaments tératogènes,…) ou certaines maladies infectieuses
(toxoplasme, virus,…) ou métaboliques (phénylcétonurie guérie, diabète,…) sont des causes
acquises bien connues de maladies congénitales malformatives et/ou de retard mental. De
même, une souffrance périnatale peut expliquer en partie ou en totalité un retard sévère des
acquisitions. En revanche, une origine génétique devra être particulièrement suspectée devant,
d’une part, la notion d’un antécédent de fausse-couches à répétition ou d’apparentes
difficultés de conception qui peuvent traduire l’existence d’une anomalie chromosomique
équilibrée chez l’un des deux parents, et, d’autre part, un âge parental élevé au moment de la
conception : âge maternel élevé favorisant les aneuploïdies (trisomie 21 notamment) et âge
paternel élevé favorisant la survenue de néo-mutations responsables d’affections dominantes
d’allure sporadique (achondroplasie par exemple). Il conviendra aussi de rechercher
l’existence d’anomalies détectées lors du suivi systématique de la grossesse et qui peuvent
être en relation avec des manifestations anténatales de l’affection: suivi échographique
principalement (biométries, clarté nucale,…) et éventuellement par le dosage au deuxième
trimestre des marqueurs hormonaux prédictif de trisomie 21.
1.1.2.2 L’enquête familiale est un moment clé de l’interrogatoire. Elle vise d’une part à
collecter les principaux antécédents médicaux des sujets apparentés au cas index et d’autre
part à préciser la structure familiale sur plusieurs générations (généralement deux ou trois). La
réalisation d’un arbre généalogique annoté est le meilleur moyen pour mettre en évidence
dans un pedigree une prédisposition héréditaire à un trait morbide et pour évoquer des modes
possibles de transmission. Ainsi, la notion dans une même fratrie (hérédité d’allure
« horizontale) de plusieurs sujets atteints d’une affection rare et ayant des parents
asymptomatiques doit orienter vers une affection transmise en récessivité. Si de plus l’analyse
de la structure familiale révèle une consanguinité parentale l’hypothèse d’une affection à
transmission autosomique et récessive devient très probable. A l’opposé, la notion de
plusieurs sujets apparentés présentant des symptômes similaires fera plutôt évoquer une
transmission en dominance si ces sujets appartiennent à des générations différentes (hérédité
d’allure « verticale »). L’impact de ces données sur le conseil génétique peut être considérable
conduisant notamment à une évaluation plus précise du risque de récurrence de l’affection
dans la famille concernée. L’arbre généalogique est donc un élément indispensable du dossier
clinique du patient. Il convient d’utiliser pour sa réalisation les symboles et règles admises
internationalement et dont les plus courants ont été représentés dans la figure ci-dessous.

5
Arbre généalogique et symboles courants
IM
I
II
III
1
2
3
4
5
1
2
3
4
5
6
7
8
9
10
1
2
3
Femme
Homme
Sexe non connu
Hétérozygote
Sujets atteints
Consultante
12SAG
Sujet décédé
Union
Divorce
Consanguinité
Pas de descendance
Propositus ou
Cas Index
12 SAG
Grossesse de 12 semaines
d’aménorrhée gravidiques
Enfant mort-né
Interruption Médicale de
Grossesse
Fausse-couche
Vrais jumeaux
III
3
Fig. 1
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
1
/
49
100%