Syndrome de Buschke–Ollendorff

Disponible en ligne sur www.sciencedirect.com
Revue du Rhumatisme 75 (2008) 292–294
Fait clinique
Syndrome de Buschke–Ollendorff夽
Buschke–Ollendorff syndrome
Hasna Hassikou∗, Fatima Tabache, Soumaya Safi, Mohamed Baaj, Larbi Hadri
Service de médecine interne, hôpital militaire Moulay-Ismail, Meknès, Maroc
Rec¸u le 8 novembre 2006 ; accepté le 3 avril 2007
Disponible sur Internet le 6 septembre 2007
Résumé
Le syndrome de Buschke–Ollendorff (SBO) est une affection autosomique dominante associant un hamartome de type élastique et une
ostéopœcilie.
Observation. – Une jeune femme âgée de 21 ans est atteinte de syndrome de Buschke–Ollendorff révélé par une ostéopœcilie. Elle présentait des
lésions cutanées à type de papules groupées de couleur chair, non douloureuses, non prurigineuses et inesthétiques au niveau de la cuisse et du
tronc. La biopsie cutanée d’une papule a montré des fibres de collagène épaissies et homogènes. Les fibres élastiques étaient larges et entrecroisées
sans augmentation de leur nombre.
Discussion. – Le syndrome de Buschke–Ollendorff est une maladie rare (un cas sur 20 000). Son diagnostic est basé sur un bon examen clinique et
sur l’analyse radiologique. Il est important d’établir le diagnostic avec précision afin d’éviter les erreurs diagnostiques (métastases condensantes)
notamment quand l’ostéopœcilie est révélatrice.
© 2008 Publi´
e par Elsevier Masson SAS.
Mots clés : Syndrome de Buschke–Ollendorff ; Ostéopœcilie
Keywords: Buschke–Ollendorff syndrome; Osteopoikilosis
1. Introduction
Le syndrome de Buschke–Ollendorff (SBO) est une affection
héréditaire rare associant un hamartome de type élastique et une
ostéopœcilie. Les progrès de la génétique ont permis de mettre en
évidence chez les individus atteints d’ostéopœcilie avec ou sans
syndrome de Buschke–Ollendorff une mutation hétérozygote,
très probablement inactivatrice, du gène LEMD3 qui code pour
une protéine de la membrane nucléaire interne. Nous rapportons
un nouveau cas de ce syndrome.
2. Observation
Une jeune femme âgée de 21 ans sans antécédents patho-
logiques est venue consulter aux urgences pour une entorse de
夽Ne pas utiliser, pour citation, la référence franc¸aise de cet article, mais sa
référence anglaise dans le même volume de Joint Bone Spine.
∗Auteur correspondant.
Adresse e-mail : [email protected] (H. Hassikou).
la cheville gauche. Cette dernière était gonflée. La radiogra-
phie des chevilles montrait des îlots de condensation arrondis de
quelques millimètres de diamètre séparés les uns des autres sur le
tarse, les métatarses et les phalanges de fac¸on bilatérale. Aucune
fracture n’était constatée. La patiente a rec¸u un traitement
symptomatique à base d’antalgiques et d’anti-inflammatoires
non stéroïdiens et a été orientée en rhumatologie pour une
enquête étiologique. L’examen clinique montrait une patiente
en bon état général pesant 58 kg pour 1,60 m. À l’examen rhu-
matologique, on ne remarquait ni douleurs ni déformations
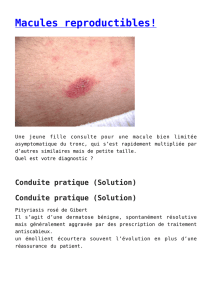
osseuses. L’examen cutané révélait à la cuisse droite (Fig. 1)
et au tronc, des papules multiples disposées en groupe à surface
irrégulière, de couleur chair, non douloureuses et non pruri-
gineuses. Ces lésions étaient présentes depuis l’enfance et se
sont étendues progressivement avec la croissance. Les examens
biologiques comprenant la vitesse de sédimentation, la numé-
ration formule sanguine, le bilan phosphocalcique, hépatique
et rénal étaient sans anomalie. La radiographie du squelette
montrait l’atteinte de tous les os (région métaphyso-épiphysaire
des os longs et du bassin (Fig. 2) à l’exception du rachis et
1169-8330/$ – see front matter © 2008 Publi´
e par Elsevier Masson SAS.
doi:10.1016/j.rhum.2007.04.018

H. Hassikou et al. / Revue du Rhumatisme 75 (2008) 292–294 293
Fig. 1. Papules groupées à la cuisse de couleur chair, à surface irrégulière.
du crâne). La biopsie cutanée de la cuisse droite avait mon-
tré des fibres de collagène épaissies et homogènes au niveau
du derme ; les fibres élastiques étaient larges et entrecroisées
sans modifications de leur structure au microscope électro-
nique. Le diagnostic d’ostéopœcilie a été porté sur la base des
données radiologiques. Son association avec les lésions cuta-
nées décrites ci-dessus permet de diagnostiquer un syndrome
Buschke–Ollendorff. Aucune étude génétique n’a pu être réali-
sée.
Fig. 2. Radiographie standard antéropostérieure du bassin révélant de multiples
zones denses arrondies et ovalaires des extrémités supérieures fémorales et du
bassin.
3. Discussion
Le SBO est une affection héréditaire rare (un cas sur 20 000
dans la population) transmise selon un mode autosomique domi-
nant à expressivité variable. Il ne connaît pas de prédilection
raciale ni de prédominance de sexe. Buschke et Ollendorff
l’ont décrit en 1928 [1]. Ce syndrome est caractérisé par la
présence d’hamartomes disséminés de type élastique (derma-
tofibrose lenticulaire disséminée) associé à une ostéopœcilie
[2,3]. Deux différentes présentations cliniques ont été décrites
dans la littérature au cours du SBO [4]. Pour la première,
il s’agit de petites papules lichénoïdes asymptomatiques, uni-
formes et symétriques. Pour la seconde présentation, les lésions
ressemblent au pseudoxanthoma elasticum. Le terme de der-
matofibrose lenticulaire disséminée est utilisé pour désigner ce
premier type. Fréquemment, les lésions sont à type de nodules
larges groupés de couleur chair et asymétriques. Cette présenta-
tion a été observée chez cette patiente et chez les trois membres
de la famille japonaise [3]. Les lésions cutanées peuvent appa-
raître à n’importe quel âge, mais sont fréquentes avant la puberté
[5]. Il ya un seul cas rapporté dans la littérature où les lésions
sont apparues à la cinquième décennie [6]. Chez cette patiente,
les lésions cutanées existaient depuis l’enfance. Les résultats de
la biopsie cutanée sont variables [3] :
•les fibres de collagène peuvent être épaissies, groupées ou
normales ;
•les fibres élastiques, quant à elles, peuvent être nombreuses
ou en faible quantité, larges et entrecroisées, fragmentées ou
normales.
La physiopathologie des lésions cutanées demeure impré-
cise. Giro et al. [7] ont constaté que les fibroblastes en culture
des patients atteints de SBO produisent trois à huit fois plus de
tropoélastine que les fibroblastes des sujets sains et que le taux
d’ARNm codant pour l’élastine était élevé. Ces signes cutanés
ne nécessitent aucun traitement spécifique.
Le SBO est associé à une ostéopœcilie. Il s’agit d’une ostéo-
pathie condensante rare qui obéit à un mode de transmission
autosomique dominant et dont le diagnostic est radiologique
[8,9]. Classiquement, les sujets atteints sont asymptomatiques,
mais 15 à 20 % décrivent des douleurs et des épanchements arti-
culaires. Chez cette malade, elle a été découverte fortuitement
suite à la radiographie réalisée pour une entorse de la cheville. La
radiographie standard montre des petites condensations squelet-
tiques arrondies ou ovalaires allant de quelques millimètres à un
centimètre de diamètre, aux contours pas toujours nets, séparés
les unes des autres. Ces lésions sont bilatérales et symétriques ;
elles sont disséminées sur les épiphyses et les métaphyses des
os long, l’os spongieux des ceintures, des os du carpe et du
tarse [10,11]. Le crâne et le rachis sont respectés. Ces lésions
n’ont pas de caractère évolutif, ne fixent pas à la scintigraphie et
n’ont aucun retentissement négatif sur la consolidation osseuse
en cas de fracture [11,12]. Les différentiels radiologiques sont
surtout l’ostéopathie striée, la mélorhéostose, la sclérose tubé-
reuse, les métastases osseuses condensantes et l’ostéome [9,10].

294 H. Hassikou et al. / Revue du Rhumatisme 75 (2008) 292–294
Chez cette malade, le diagnostic de SBO n’a pu être établi que
devant la découverte fortuite des images d’ostéopœcilie. Les
lésions cutanées étaient négligées car elles n’avaient entraîné
aucune douleur ni gêne esthétique. Une étude génétique menée
par les méthodes classiques d’analyse de liaison sur l’ensemble
du génome a été menée sur trois familles avec ostéopœcilie
avec ou sans syndrome de Buschke–Ollendorff ou mélorhéos-
tose. Tous les individus atteints étaient porteurs d’une mutation
hétérozygote, très probablement inactivatrice, du gène LEMD3
(également appelé MAN1) qui code pour une protéine de la
membrane nucléaire interne. Chez les quelques patients pour
lesquels cette recherche a été effectuée, une mutation somatique
du deuxième allèle de LEMD3 n’a pu être mise en évidence que
ce soit sur les fibroblastes de la peau atteinte dans un cas de
syndrome de Buschke–Ollendorff ou sur la peau apparemment
normale d’un patient atteint de mélorhéostose, ce qui laisse pen-
ser que la perte de fonction de 50 % de la molécule suffit à faire
apparaître le phénotype pathologique, un phénomène connu sous
le nom d’haplo-insuffisance. La fonction de la protéine codée
par le gène LEMD3 n’est pas encore clairement identifiée, mais
elle semble interagir en inhibant les deux voies de signalisation
intracellulaire médiées par BMP et SMADS ; cette dernière voie
dépendant notamment de l’activation des récepteurs au TGFB
et à l’activine. [13–15]. Le SBO ne semble pas entraîner une
augmentation de la morbidité ou de la mortalité, sauf dans les
rares cas de dégénérescence maligne des lésions osseuses [16].
Références
[1] Buschke A, Ollendorff H. Ein fall von dermatofibrosis lenticularis dis-
seminata und osteopatha condensans disseminata. Derm Wochenschr
1928;86:257–62.
[2] Woodrow SL, Pope FM, Handfield-Jones SE. The Buschke–Ollendorff
syndrome presenting as familial elastic tissue naevi. Br J Dermatol
2001;144:890–3.
[3] Kawamura A, Ochiai T, Tan-Kinoshita M, et al. Buschke–Ollendorff
syndrome: three generations in a japanese family. Pediatr Dermatol
2005;22:133–7.
[4] Foo CC, Kumarasinghe SP. Juvenile elastoma: a form fruste of
the Buschke–Ollendorff syndrome ? Australas J Dermatol 2005;46:
250–2.
[5] Cairns RJ. Familial juvenile elastoma: osteopoikilosis (two cases). Proc R
Soc Med 1967;60:1267.
[6] Reinhardt LA, Rountree CB, Wilkin JK. Buschke–Ollendorff syndrome.
Cutis 1983;31:94–6.
[7] Giro MG, Duvic M, Smith LT, et al. Buschke–Ollendorff syndrome associa-
ted with elevated elastin production by affected skin fibroblasts in culture.
J Invest Dermatol 1992;99:129–37.
[8] Carvalho AC, Santos Beze R, Picinini SE. Osteopoikilosis – a case report
and review of the literature. Radiol Bras 2002;35:1–6.
[9] Borman P, Özoran K. Ostéopoecilie : un cas clinique et revue de la littéra-
ture. Rev Rhum 2002;69:326–30.
[10] Kotulska A, Kucharz EJ. Osteopoikilosis and Buschke–Ollendorff syn-
drome. Case Rep Clin Pract Rev 2002;3:290–3.
[11] Zahar A, Najeb Y, Rafai M, et al. Fracture du col du fémur sur ostéopœcilie.
Rev Chir Orthop Réparatrice Appar Mot 2002;88:725–7.
[12] Buyukbebeci O, Karakurum G, Sirikc¸i A, et al. Fracture healing in a patient
with osteopoikilosis. Joint Bone Spine 2005;72:343–4.
[13] Dereure O. Buschke–Ollendorff syndrome: inactivating mutation of the
LEMD3 gene. Ann Dermatol Venereol 2005;132:593.
[14] Lin F, Morrison JM, Wu W, et al. MAN1, an integral protein of the
inner nuclear membrane, binds Smad2 and Smad3 and antagonizes
transforming growth factor-beta signaling. Hum Mol Genet 2005;14:
437–45.
[15] Hellemans J, Debeer P, Wright M, et al. Germline LEMD3 mutations
are rare in sporadic patients with isolated melorheostosis. Hum Mutat
2006;27:290.
[16] Grimer RJ, Davies AM, Starkie CM, et al. Chondrosarcoma in a patient
with osteopoikilosis. À propos of a case. Rev Chir Orthop Reparatrice
Appar Mot 1989;75:188–90.
1
/
3
100%