Chimie organique

ENSEIGNEMENT DE PROMOTION SOCIALE
——————————————————————
Cours de
CHIMIE ORGANIQUE
- Alcènes -
——————————————————————
H. Schyns
Décembre 2006

AlcènesSommaire
H. SchynsS.1
Sommaire
1.PRÉSENTATION GÉNÉRALE
1.1.Structure générale
1.2.Nomenclature
1.3.Stéréoisomérie
1.4.Conjugaison et délocalisation
1.5.Propriétés physiques
1.5.1.Forme physique
1.5.2.Densité
1.5.3.Solubilité
1.6.Réactivité
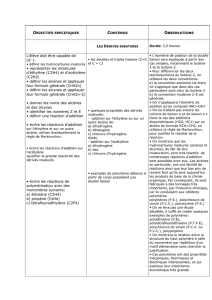
2.RÉACTIONS DES ALCÈNES
2.1.Aperçu
2.2.Additions
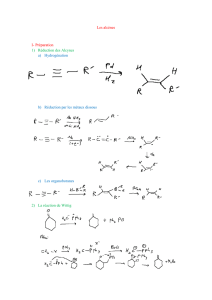
2.2.1.Addition catalytique d'hydrogène
2.2.2.Principe des additions électrophiles
2.2.3.Addition d'hydracides halogénés (Markownikov)
2.2.4.Addition d'hydracides halogénés (anti-Markownikov)
2.2.5.Addition d'eau (Markownikov)
2.2.6.Hydratation par hydroboration (anti-Markownikov)
2.2.7.Addition d'acides hypohalogénés
2.2.8.Addition de dihalogènes
2.3.Substitution en α
2.3.1.Substitution radicalaire par un halogène
2.4.Oxydations
2.4.1.Degrés d'oxydation et oxydants
2.4.2.Epoxydation par peracide
2.4.3.Cis-hydroxylation par KMnO4
2.4.4.Ozonolyse
2.4.5.Coupure oxydative par un oxydant fort
2.5.Polymérisation
2.5.1.Principe de la polymérisation
2.5.2.Polymérisation radicalaire
2.5.2.1En bref
2.5.2.2Phase d'initiation
2.5.2.3Phase de propagation
2.5.2.4Phase de terminaison
2.5.3.Polymérisation cationique
2.5.3.1En bref
2.5.3.2Phase d'initiation
2.5.3.3Phase de propagation
2.5.3.4Phase de terminaison

AlcènesSommaire
H. SchynsS.2
3.PRODUCTION DES ALCÈNES
3.1.A partir des dérivés halogénés
3.1.1.Par élimination d'un monohalogéné
3.1.2.Par élimination d'un dihalogéné vicinal
3.2.A partir des alcools, par élimination
3.3.A partir des alcynes, par hydrogénation
4.RÉSUMÉ
4.1.Production
5.NOTE SUR LA CATALYSE
6.SOURCES

Alcènes1 - Présentation générale
H. Schyns1.1
1. Présentation générale
1.1. Structure générale
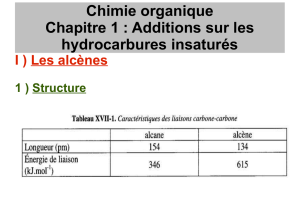
Comme les alcanes, les alcènes sont composés exclusivement de carbone et
d'hydrogène. Contrairement aux alcanes, qui ne contiennent que des liaisons
simples, les alcènes contiennent au moins une liaison double entre deux atomes
de carbone :
fig. 1.1 Un alcène linéaire, ramifié et cyclique
Ils peuvent de présenter sous une forme linéaire, ramifiée ou cyclique.
Par rapport aux alcanes, les alcènes ont dû perdre deux atomes d'hydrogène afin de
pouvoir former la double liaison. La formule brute des formes linéaires ou
ramifiées est donc :
CnH2n
Les cycloalcènes ont dû perdre deux atomes d'hydrogène de plus afin de pouvoir
fermer le cycle :
CnH2n-2
La double liaison implique que deux atomes de carbone adjacents sont hybridés
sous la forme sp2 .La double liaison est composée d'une liaison σ obtenue par
recouvrement des orbitales hybridées, et d'une liaison π, obtenue par mise en
parallèle et recouvrement des orbitales 2p non hybridées.
Rappelons qu'un atome de carbone qui est impliqué dans deux doubles liaisons est
hybridé sous la forme sp. Dans l'exemple de la fig. 1.2, le carbone central est
hybridé sp, ses voisins sont hybridés sp2 et ceux des extrémités sont hybridés sp3.
CH3
CCC
CH3
H
H
fig. 1.2 Un atome de carbone sp porteur de deux doubles liaisons
1.2. Nomenclature
La nomenclature des alcènes a été abordée dans la partie intitulée "Aperçu général
et Nomenclature".
Le nom de l'alcène dérive de celui de l'alcane correspondant en remplaçant
la terminaison -ane par -ène.
Pour indiquer la position de la double liaison dans cette chaîne, on fait précéder le
suffixe -ène par le numéro du premier carbone qui y est impliqué. La numérotation
est faite de manière à donner le plus petit numéro à la position de la double liaison :

Alcènes1 - Présentation générale
H. Schyns1.2
CH31C
H
2
C
H
3CH2
4
CH2
5CH3
6
fig. 1.3 Hex-2-ène
Si la chaîne est ramifiée, la chaîne principale à considérer n'est pas la plus
longue mais bien la plus longue contenant la double liaison.
De plus, la double liaison a priorité sur tous les substituants, c'est elle qui impose
le sens de numérotation :
12
345678
fig. 1.4 7-Methyl-3-propyloct-2-ène
Quand l'hydrocarbure présente plusieurs doubles liaisons, on insère les préfixes di,
tri, etc dans la terminaison :
fig. 1.5 Penta-1,3-diène
Quelques alcènes possèdent des noms usuels qui s'écartent de la règle IUPAC :
CH2CH2Ethylène
CH2CH
CH3
Propylène
1.3. Stéréoisomérie
A cause de l'hybridation sp2, les six atomes qui entourent la double liaison sont dans
le même plan. La double liaison est rigide; elle interdit la libre rotation des atomes
de carbone autour de l'axe C=C.
De ce fait, si les quatre valences libres sont occupées par des groupes différents
(R1, R2, R3, R4), on peut concevoir six molécules différentes qui ne diffèrent que par
l'agencement des groupes autour de la double liaison :
Cis
ou
Z
R1
R2
R3
R4
R1
R3
R2
R4
R1
R4
R2
R3
Trans
ou
E
R1
R2
R4
R3
R1
R3
R4
R2
R1
R4
R3
R2
fig. 1.6 Stéréoisomères et isomères de squelette
Pour illustrer ce constat, admettons que R1
, R2
, R3
, R4
soient respectivement des
chaînes linéaires à 1, 2, 3, 4 atomes de carbone.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
1
/
32
100%