Poly CPCP - Mort subite du nourrisson - polys-ENC

C@mpus National de pédiatrie et chirurgie pédiatrique TICEM – UMVF
Auteur : Pr. Gérard Chéron MAJ : 06/01/2005
Dr Timsit Sandra ( Hôpital Necker Enfants Malades – Paris)
Mort Subite – Item 210
Objectifs :
OBJECTIFS OFFICIELS de l’ECN
Expliquer la définition de la mort subite du nourrisson, son épidémiologie, les facteurs
de risque et de prévention , et les principes de la prise en charge de la famille.
OBJECTIFS du Collège des professeurs de pédiatrie (5.8)
OBJECTIFS TERMINAUX
Exposer la définition, l’épidémiologie de la mort subite, les hypothèses étiologiques,
les facteurs de risque et les principes de la prise en charge de la famille d’un enfant
mort de mort subite.
OBJECTIFS PEDAGOGIQUES INTERMEDIAIRES
¤ Définir la mort subite du nourrisson, citer les données épidémiologiques et décrire
les signes d’alarme prémonitoires.
¤ Décrire les causes et mécanismes possibles d’une mort subite du nourrisson.
¤ Décrire la conduite à tenir par le médecin qui constate une mort subite inopinée
chez un nourrisson.
¤ Décrire et justifier les recommandations actuelles de couchage du nourrisson.
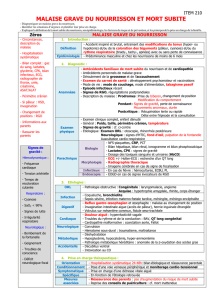
Afin d’éviter toute confusion entre ces deux situations, la question a été scindée en
deux parties : tout d’abord malaise grave du nourrisson puis mort subite.
Sommaire
Défintion
Faq 1 - Epidémiologie
Faq 2 - Clinique
Faq 3 - Conduite à tenir
Faq 4 - Autopsie
Faq 5 - Résultats
Faq 6 - Aspects étiologiques potentiels des MSIN
Faq 7 - Prévention
Définition
La mort subite du nourrisson (MSN) est le décès brutal et inattendu d’un enfant
considéré jusque-là comme bien portant, décès qu’aucun examen, y compris
l’autopsie, ne peut expliquer. Cette définition internationale a été reprise par le code
OMS des maladies.
1

C@mpus National de pédiatrie et chirurgie pédiatrique TICEM – UMVF
Auteur : Pr. Gérard Chéron MAJ : 06/01/2005
Dr Timsit Sandra ( Hôpital Necker Enfants Malades – Paris)
Faq 1 - Epidémiologie
Facteurs de risque. En France, 0,7‰ des nourrissons décèdent soudainement au
berceau. Le sexe masculin, un âge inférieur à 6 mois (95%), la recrudescence
hivernale, la prématurité, le retard de croissance intra-utérin ou une pathologie
néonatale grave sont les principales caractéristiques épidémiologiques de ces morts
subites. Le risque est majoré chez les enfants de mère jeune, vivant dans un
contexte socio-économique défavorisé, tabagique ou droguée. Il existe enfin des
familles touchées par plusieurs décès au sein d’une même fratrie. Elles témoignent
très probablement d’un terrain particulier ou d’une maladie héréditaire (neurologique,
métabolique, musculaire…) non diagnostiquée chez les enfants décédés.
Faq 2 - Clinique
L’accident est stéréotypé. C’est un nourrisson de 2 à 4 mois trouvé mort dans son
berceau, couché sur le ventre, à l’heure habituelle d’un biberon. Les gestes de
réanimation sont inefficaces.
En pratique, le diagnostic de MSN doit être porté au terme d’un bilan post-mortem. Il
est donc indispensable de rechercher les facteurs favorisants, les circonstances et
de réaliser une véritable consultation pédiatrique. Ceci impose d’accueillir la famille,
de recueillir progressivement à l’occasion d’entrevues répétées, l’histoire familiale et
personnelle de l’enfant, de réaliser un examen clinique complet, des examens
complémentaires biologiques, microbiologiques, radiologiques et d’obtenir des deux
parents l’autorisation écrite d’autopsie. Une étiologie précise au décès du nourrisson
est un point de départ important pour le travail de deuil. C’est aussi la condition d’une
prise en charge adaptée et objective de tout nouvel enfant de ce couple.
Faq 3 - Conduite à tenir
L’examen clinique est complet : aspect morphologique, cutanéo-muqueux, état
trophique, prise de la température rectale, état d’hydratation, existence de sueurs,
d’une pâleur, d’une cyanose, de lividités, d’une odeur particulière, d’une rigidité
cadavérique, de rejet nasal ou buccal, palpation des segments de membres, de
l’abdomen, examen de la cavité buccale, mesure du poids, de la taille, du périmètre
crânien.
Les examens complémentaires sont tous possibles. Tous sont importants :
radiographie de thorax et du squelette, numération formule sanguine, réticulocytes,
hémoculture, prélèvements microbiologiques : pharyngé, trachéal, ponction lombaire
(chimie, cytologie, bactériologie, virologie), ponction péricardique et pleurale à la
recherche d’un épanchement (bactériologie, virologie, cytologie), recueil d’urines et
de sang pour une étude métabolique.
Les parents sont reçus par le pédiatre pour connaître les circonstances du décès,
s’informer des antécédents familiaux et personnels, consulter le carnet de santé de
l’enfant et aborder la question de l’autopsie. Son utilité est indiscutable mais il est
difficile de convaincre sans heurter. Aucune législation, aucune religion ne s’oppose
à cet examen.
2

C@mpus National de pédiatrie et chirurgie pédiatrique TICEM – UMVF
Auteur : Pr. Gérard Chéron MAJ : 06/01/2005
Dr Timsit Sandra ( Hôpital Necker Enfants Malades – Paris)
Faq 4 - Autopsie
Les renseignements obtenus dépendent de la précocité et de la méthodologie de
l’autopsie. L’étude macroscopique est complète, thoracique, abdominale, le bloc ORL
mais aussi l’encéphale, le cervelet, le tronc cérébral, les adénopathies cervicales. En
cours d’autopsie sont effectués des prélèvements stériles pour des recherches de
virus en culture de tissu (encéphale, poumon, cœur, foie).
Chaque organe est examiné, pesé, mesuré à la recherche d’une lésion, d’une
malformation, puis il fait l’objet de plusieurs coupes, selon des repères anatomiques
précis, pour l’étude histologique.
Faq 5 - Résultats
L’ensemble du dossier est discuté avec tous les membres de l’équipe : métabolicien,
microbiologiste, hématologiste, radiologue, les médecins du secours d’urgence
appelés au domicile et l’anatomo-pathologiste.
Trois situations sont possibles :
– Le décès est rattaché à une cause précise parce qu’il y a une parfaite concordance
anatomo-clinique. L’autopsie a découvert une malformation cardiaque grave, une
myocardite, une myocardiopathie, une infection aiguë multiviscérale, une tumeur
cardiaque, une hypoplasie congénitale du cortex surrénalien, une méningo-
encéphalite, les stigmates de mauvais traitement, un rachitisme carentiel massif…
Cette situation est fréquente dans les décès précoces du fait de l’importance des
infections bactériennes et virales le premier mois de vie.
– Le décès a une cause possible en raison de lésions témoignant d’une pathologie
récente ou chronique, méconnue avant le décès. Il s’agit de lésions siégeant sur un
seul organe ou un seul système, très fréquemment l’arbre respiratoire (laryngite,
épiglottite, infection bronchique ou pulmonaire). La découverte d’un élément
microbiologique est importante pour valider le caractère pathogène des lésions.
– Le décès ne reçoit aucune explication : il s’agit d’une mort subite inexpliquée du
nourrisson (MSIN). Il est probable que ce cadre sera démembré au prix de nouveaux
progrès méthodologiques dans le bilan post-mortem.
Faq 6 - Aspects étiologiques potentiels des MSIN
Ces morts au berceau relèvent de pathologies multifactorielles. Plusieurs
événements se succèdent, s’associent, s’amplifient.
– La recrudescence hivernale, la présence d’une rhinopharyngite avant le décès, les
lésions inflammatoires du système respiratoire, la découverte de virus dans les
lésions sont des facteurs bien documentés. Des médiateurs de l’inflammation
(interféron, interleukines) sont présents dans le liquide céphalo-rachidien et le
plasma. Des enzymes du métabolisme de détoxification des xénobiotiques
(cytochromes) sont anormalement représentés.
– L’hyperthermie et des variations brusques et importantes de la température
centrale (hyper ou hypothermie) modifient la régulation respiratoire. Les
hyperthermies ne sont pas toutes infectieuses. Elles peuvent être exogènes : enfant
trop couvert, trop près d’une source de chaleur. Elles créent des pertes d’eau
importantes, une déshydratation.
– Le reflux gastro-œsophagien peut être cause d’une inhalation alimentaire massive.
3

C@mpus National de pédiatrie et chirurgie pédiatrique TICEM – UMVF
Auteur : Pr. Gérard Chéron MAJ : 06/01/2005
Dr Timsit Sandra ( Hôpital Necker Enfants Malades – Paris)
Celle-ci est rare. Ces reflux se compliquent aussi de malaise et de perte de
connaissance documentés par des enregistrements cardiaques et respiratoires. Avec
ou sans œsophagite, le reflux est, dans certains cas, cause d’un réflexe vagal
bradycardisant ou apnéisant. Il n’a néanmoins pas été démontré qu’il peut à lui seul
et en l’absence de fausse route massive, être responsable de décès.
– Des apnées centrales ont été évoquées. Un nourrisson né à terme, eutrophique,
sans pathologie néonatale, n’a pas une respiration régulière et il fait des pauses
respiratoires d’origine centrale de brève durée (< 15 s). Ce phénomène est
physiologique et en aucun cas prédictif d’un accident de MSN.
La responsabilité des apnées est encore débattue pour les prématurés, les nouveau-
nés hypotrophiques ou encore pour les enfants ayant présenté une grande
souffrance pernatale justifiant des mesures de réanimation. C’est dans ce cadre bien
particulier que des lésions anatomiques du tronc cérébral (séquelles d’infection,
d’accidents vasculaires ou d’hypoxie) peuvent être responsables d’apnées centrales
anormalement longues (> 20 s) et potentiellement pathologiques (apnée puis
bradycardie).
– Les “apnées” obstructives sont connues lors des anomalies malformatives de la
filière laryngo-pharyngée (syndrome de Pierre-Robin, laryngomalacie, rétrécissement
des voies aériennes supérieures) ou lors des problèmes infectieux (laryngite, rhinite,
épiglottite) ou chimique (irritation des reflux gastro-œsophagiens graves).
– Les troubles du rythme cardiaque sont rares. Si le syndrome du QT long, avec ou
sans surdité, est exceptionnel chez le nourrisson, les autres troubles du rythme
(tachycardie supra ventriculaire ou jonctionnelle, bloc auriculo-ventriculaire) doivent
être dépistés dès la période néonatale. Ils sont responsables d’accès de pâleur,
d’adynamie, de brève perte de contact voire d’accès de cyanose ou lorsqu’ils se
prolongent de l’installation d’une insuffisance cardiaque.
– Le syndrome de Silverman doit systématiquement être évoqué mais il faut insister
sur la très grande prudence avec laquelle il convient de chercher les stigmates
cutanés, muqueux, osseux, nutritionnels, psychiatriques et sociaux qui étayent ce
diagnostic.
– Des anomalies héréditaires du métabolisme des acides gras ont été documentées.
Les acides gras deviennent, en cas de jeûne prolongé, l’aliment énergétique en
remplacement du glucose. Leur utilisation nécessite des déshydrogénations
successives pour fournir des acyl COA. Des déficits en acyl COA déshydrogénase (à
chaîne longue, moyenne ou courte) ou des déficits en carnitine (cofacteurs des
déshydrogénases) provoquent une symptomatologie bruyante à type de malaise
hypoglycémique sans cétose, d’encéphalopathie, de syndrome de Reye, de coma,
d’acidose métabolique sévère… Ces manifestations surviennent plus volontiers à
l’occasion d’un stress (infections intercurrentes, jeûne prolongé…) qui augmente les
besoins énergétiques. Seul le dosage de l’activité enzymatique sur lymphocytes frais
ou sur culture de fibroblastes permet le diagnostic.
Faq 7- Prévention
La difficulté réside dans la prévention de tout ou partie de ces décès dont une faible
part est précédée de symptômes peu ou pas spécifiques. Il est nécessaire d’être
vigilant lors de chaque examen des nourrissons, de tenir compte des antécédents
anténataux et/ou périnatals (souffrance, réanimation, prématurité, hypotrophie), de
vérifier le développement staturo-pondéral des premiers mois en s’aidant des
4

C@mpus National de pédiatrie et chirurgie pédiatrique TICEM – UMVF
Auteur : Pr. Gérard Chéron MAJ : 06/01/2005
Dr Timsit Sandra ( Hôpital Necker Enfants Malades – Paris)
5
courbes de croissance, de penser systématiquement à l’insuffisance ou au caractère
limité des stocks vitaminiques et calciques des enfants nés prématurément,
hypotrophes ou de mères n’ayant pas bénéficié d’un ensoleillement suffisant au
cours de leur grossesse, de prendre le temps d’expliquer son ordonnance et de
chercher à savoir si elle a été comprise.
Les résultats des campagnes médiatiques sur la position dans laquelle coucher les
nourrissons sont spectaculaires et ils posent de nombreuses questions. Sur tous les
continents, une baisse de 40 à 50% des morts subites est enregistrée presque
simultanément aux messages diffusés pour encourager le décubitus dorsal. Le
décubitus ventral diminuerait les possibilités d’échange thermique, créerait les
conditions d’une ascension de la température corporelle. Une infection intercurrente
même bénigne, un excès de température majorerait alors le risque d’hyperthermie.
Couché sur le ventre, le nourrisson risque d’autre part de ventiler dans un micro
environnement enrichi en gaz carbonique, appauvri en oxygène. En conséquence, si
les nourrissons ayant un important reflux gastro-œsophagien non maîtrisé par un
traitement médicamenteux et ceux porteurs d’anomalies crânio-faciales doivent être
maintenus en orthostatisme ventral, il convient d’inciter tous les parents dès la
maternité à coucher le nouveau-né sur le dos, à ne pas chauffer exagérément sa
chambre, à ne pas trop le couvrir, à proscrire l’usage des draps, couettes,
couvertures, oreillers et tours de lit sous lesquels les nourrissons risquent de se
trouver enfouis.
1
/
5
100%