Formes familiales des cancers de l`appareil urogénital

ARTICLE DE REVUE Progrès en Urologie (1996), 6, 343-355
343
Formes familiales des cancers de l’appareil urogénital :
Aspects cliniques et génétiques
Georges FOURNIER (1), Antoine VALERI (1), Olivier CUSSENOT (2)
(1) Service d’Urologie, Hôpital Morvan, Brest, (2) Service d’Urologie, Hôpital Saint-Louis, Paris
RESUME
Les formes familiales des cancers du rein, de la voie
excrétrice et du testicule sont rares (1 à 2%),
contrairement au cancer de la prostate (20%). Au
sein de ces cancers familiaux, les formes hérédi-
t a i res en rapport avec une anomalie génétique
transmise à la descendance sont actuellement mieux
connues et ont un intérêt particulier pour le clini-
cien. Leur diagnostic peut modifier les modalités de
traitement du patient atteint compte tenu de la mul-
tifocalité des tumeurs au sein du même organe et/ou
de la bilatéralité fréquente en cas d’organes pairs.
Le risque de transmission du gène délétère à la des-
cendance conduit à une information et une sur-
veillance étroite des apparentés pour un diagnostic
précoce et un meilleur pronostic.
Lorsque le gène de prédisposition est connu, il est
possible de cibler la surveillance uniquement sur les
sujets porteurs du gène délétère compte tenu du
risque accru de cancer par rapport à la population
générale: c’est le cas pour le cancer du rein au cours
de la maladie de Von Hippel Lindau, le néphroblas-
tome et les exceptionnelles tumeurs de la voie excré-
trice dans le syndrome de Lynch. Pour le cancer de
la prostate, le plus fréquent, où les formes hérédi-
taires représenteraient 9% des cas, le gène prédis-
posant n’est pas identifié et doit conduire actuelle-
ment à proposer un dépistage chez tous les hommes
de la famille à partir de 40 ans compte tenu de l’âge
plus précoce de survenue.
Mots-clés : Cancer uro-génital, hérédité, génétique, dépistage,
conseil génétique.
Progrès en Urologie (1996), 6, 343-355.
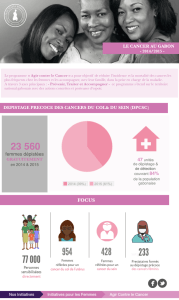
Les formes familiales représentent 5 à 10% des cancers
en général [27] et 1 à 2% des cancers de l’appareil uri-
naire et génital masculin, cancer de la prostate exclu.
Pour ce dernier, les formes familiales sont estimées à
20% des cas et 5 à 10% seraient héréditaires, comme
pour les cancers du sein et du côlon [13].
Au sein des formes familiales de cancers, on distingue
les formes héréditaires en rapport avec une anomalie
d’un (ou de plusieurs) gène(s) transmissibles au cours
des générations, et les formes familiales non hérédi-
taires qui peuvent être la conséquence de l’exposition
de la famille à un carcinogène extérieur ou le fait du
hasard (*).
Le diagnostic des formes héréditaires peut être suspec-
té par l’enquête généalogique et les caractéristiques
propres des tumeurs. En effet ces cancers ont en com-
mun un âge de survenue généralement plus précoce
que les formes sporadiques et/ou des localisations mul-
tiples primitives (multifocalité des tumeurs au sein du
même organe, ou coexistence de tumeurs d’organes
différents) [7, 27, 47]. Le support génétique de ces
formes héréditaires a été proposé initialement par
KNUDSON [48, 49] (Figure 1).
La reconnaissance de ces formes héréditaires par le cli-
nicien est essentielle car elle a des conséquences pra-
tiques pour la prise en charge du patient atteint et de ses
apparentés:
• Pour le patient le traitement du cancer peut être diffé-
rent des formes sporadiques du fait de la bilatéralité
fréquente en cas d’organes pairs.
• Pour la famille la surveillance des apparentés est néces-
saire afin de détecter précocément les cancers et ainsi
améliorer le pronostic. Dans les cancers où l’anomalie
d’un chromosome (anomalie du caryotype) ou d’un gène
spécifique est déjà identifiée il est possible de distinguer
dans la famille les membres porteurs de l’anomalie qui
bénéficieront du dépistage de ceux, indemnes dont le
risque de cancer n’est pas supérieur à celui de la popula-
tion générale. Ces notions ont conduit au concept de
conseil génétique en cancérologie(*). Ce diagnostic
génétique (et «prédictif») est déjà possible pour les can-
cers du côlon, du sein et de la thyroïde [18, 39].
Cette revue concernant les formes familiales des can-
Manuscrit reçu le 2 janvier 1996, accepté : février 1996.
Adresse pour correspondance : Dr.G.Fournier, Service d’Urologie, Hôpital de la
Cavale Blanche, Brest Cedex.
(*) Pour les lecteurs pour lesquels la tcrminologie génétique
n’est pas familière, un glossaire a été inclus à la suite du texte.
Les termes explicités dans ce glossaire sont indiqués dans le
texte par un astérisque (*).

344
cers urogénitaux a pour objectifs d’exposer par organe
les caractéristiques cliniques et génétiques de ces
tumeurs et de préciser les modalités de prise en charge
des patients et de leurs apparentés au sein de ces
familles.
Les références bibliographiques de cette revue ont été
obtenues à partir des données informatisées du
«Medline» (de janvier 1983 à septembre 1995) à partir
des items «urogenital neoplasms», « familial» et «here-
ditary». Ont été exclus les articles de recherche expéri-
mentale animale. Lorsqu’une étude de revue récente
des cas publiés pour un organe donné était disponible
dans la littérature, nous n’avons pas recherché tous les
cas publiés antérieurement.
CANCER DU REIN
On distingue 2 types de cancers du rein familiaux chez
l’adulte : le cancer du rein dans le cadre de la maladie
de Von Hippel-Lindau qui est le plus fréquent, et le
cancer du rein «commun»(*), qui n’est rattaché à aucun
syndrome particulier. Enfin, chez l’enfant, environ 1%
des tumeurs de Wilms (néphroblastome) sont des
formes familiales [6].
Maladie de Von Lippel-Lindau (VHL)
Aspects génétiques
Cette phacomatose héréditaire est très rare : incidence
de 1/36000 naissances, prévalence de 1/53000 habi-
tants et 700 cas publiés [56, 73, 97] Il s’agit d’une
maladie à transmission autosomique dominante (*), à
forte pénétrance (*) (Tableau 1), pour laquelle un seul
gène est en cause, situé sur le bras court du chromoso-
me 3 (3p25-26) (*) (voir locus chromosomique) [35,
43, 55, 56, 104] (Tableau 1). La protéine correspon-
dante de siège membranaire est impliquée dans la
transduction du signal cellulaire et l’adhésion inter-cel-
lulaire [56, 97]. La variété des lésions possibles pour-
rait être expliquée par des altérations du gène de pré-
disposition (*) en des sites variables [55].
Aspects cliniques
Parmi les 25 lésions décrites dans la maladie de VHL,
le cancer du rein est au troisième rang et chaque type
de tumeur a un âge spécifique de début (Tableau 2). Un
patient peut présenter successivement plusieurs locali-
sations, par exemple une tumeur oculaire à 20 ans, une
tumeur cérébrale à 30 ans, et un cancer du rein à 40 ans
[55]. Sa forme histologique est celle habituellement
rencontrée dans les formes sporadiques (adénocarcino-
me à cellules claires) [92, 97] et la fréquence des méta-
stases n’est pas différente malgré un plus faible grade
nucléaire et une évolution longtemps locale [46,75].
Les patients atteints de VHL vivent en moyenne jus-
qu’à 50 ans [37, 56]. Le pronostic de la maladie a été
amélioré par la réduction de la mortalité due aux
hémangioblastomes cérébelleux. Il dépend actuelle-
ment de l’évolution du cancer du rein dont la survenue
est plus tardive dans l’histoire de la maladie [19, 97].
Le diagnostic de cancer du rein est effectué dans deux
circonstances :
• 1er cas : La maladie est connue et le cancer du rein
souvent asymptomatique est alors découvert par l’ima-
gerie au cours de la surveillance des membres de la
famille.
Néanmoins l’imagerie est d’interprétation difficile en
cas de lésions infracentimétriques (non détectées par la
tomodensitométrie ou l’IRM), ou quand il existe de
multiples kystes rénaux, ou encore lorsque les tumeurs
sont situées dans la paroi même des kystes [83, 92].
Des lésions dysplasiques siégeant dans la paroi des
kystes pourraient alors expliquer le développement de
telles tumeurs [16, 92, 97]. Dans la série de POSTON
[92], la fréquence des cancers du rein développés dans
des kystes est de 21% (examen anatomopathologique
de 116 lésions kystiques obtenues à partir de pièces de
néphrectomie pour cancer du rein en cas de VHL).
• 2ème cas : Quand la maladie n’est pas connue, c’est
la tumeur du rein qui est au premier plan. Certaines
caractéristiques de la tumeur, propres aux cancers à
prédisposition génétique sont évocatrices: âge de sur-
venue plus précoce, lésions fréquemment multifocales
et bilatérales, synchrones ou différées, parfois très tar-
Figure 1. Modèle de carcinogénèse développé initialement par
KNUDSON dans le rétinoblastome [48, 49].Ce modèle, confirmé
par de nombreux travaux, suggère l’existence d’altérations
génétiques similaires dans les formes sporadiques et familiales
de cancers [7, 48, 49].A la différence des formes sporadiques,
les patients atteints de formes héréditaires ont une anomalie
génétique constitutionnelle présente à la naissance et ont déjà
franchi la première étape de la carcinogénèse. Un second
modèle, différent de celui de KNUDSON, suppose l’existence
préalable d’une altération moléculaire héritée, rendant la cel -
lule sensible à des évènements oncogéniques successifs : théo -
rie du «phénotype mutateur» [63].

divement (jusqu’à 14 ans après la première localisa-
tion) [20] (Tableau 3).
L’enquête familiale et la recherche d’autres localisa-
tions de la maladie permettront dans la majorité des cas
de reconnaître la maladie de VHL. En l’absence d’an-
técédents familiaux, c’est l’existence d’au moins une
localisation supplémentaire de la maladie chez le
patient qui permet le diagnostic [97].
L’absence d’antécédents familiaux chez des sujets
atteints de la maladie de VHL peut être expliquée, en
dehors des cas de fausse paternité, par l’existence d’une
mutation « de novo », absente dans le génome parental
[55].
Implications pratiques
Patient
En l’absence de métastases, le traitement chirurgical
des tumeurs rénales, en majorité bilatérales et multifo-
cales, est controversé. Certains auteurs ont proposé une
attitude radicale d’emblée par binéphrectomie en cas de
tumeurs bilatérales compte tenu du caractère inévita-
blement récidivant de la maladie en rapport avec la pré-
disposition génétique et du risque d’apparition de méta-
stases en cas de tumeur résiduelle [28]. D’autres prô-
nent une chirurgie conservatrice (néphrectomie partiel-
le ou tumorectomies multiples) avec surveillance étroi-
te du parenchyme rénal restant, en dehors des cas où le
caractère diffus et bilatéral des lésions impose une
néphrectomie bilatérale de nécessité [64, 85, 90]. Les
arguments avancés par les défenseurs de cette attitude
sont en rapport avec les caractéristiques de ces
tumeurs : facilité d’énucléation, bas grade et stade pré-
coce, rareté des métastases au cours du suivi des
patients avec un recul variable selon les séries (6 mois
à 8 ans pour les 13 patients des 3 séries de LOUGHLIN
[64], PEARSON [90] et LEVINE [61], détérioration de la
qualité de vie des patients anéphriques en dialyse et
risque carcinogène d’un traitement immunosuppres-
seur prolongé en cas de transplantation rénale secon
-
daire [46]. En réalité, le suivi à long terme de 9 patients
dans la série de NOVICK (61 à 120 mois, 86 mois en
moyenne) montre que si un des patients est vivant sans
récidive à 74 mois, un autre est mort de métastases à 43
mois, et les 7 patients restants ont présenté une récidi-
ve sur le rein restant [85].Ces résultats ainsi que l’ex-
périence rapportée récemment par la même équipe à
propos de 5 patients transplantés avec un recul de 7 à
66 mois (4 patients vivants avec un greffon rénal fonc-
tionnel sans récidives) [110], ne permettent pas de pro-
345
Tableau 1. Aspects génétiques des cancers du rein familiaux [11, 35, 38, 42, 43, 50, 78, 97, 98, 102, 103, 108, 119].
Type de cancer Mode de transmission Pénétrance Gène(s) impliqué(s) Dépistage génétique
Cancer du rein V.H.L. Autosomique 70% à 60 ans VHL (3p 25-26) Oui
dominant
Cancer du rein commun Autosomique NP NP Non
(Tumeurs non papillaires) dominant?
--------------- --------------------------------------------------------------------------------------------------------------------------------------
Cancer du rein commun
(Tumeurs tubulo-papillaires) Autosomique NP NP Non
dominant?
Tumeurs de Wilms Autosomique variable WT1 (11p13) Oui
dominant WT2 (11p15)
WT3?
NP : non précisé.
VHL : maladie de Von Hippel-Lindau.
Tableau 2. Types de lésions en cas de maladie de Von Hippel-
Lindau [34, 46, 55, 74, 97, 98].
Atteinte Fréquence (%) Age moyen de survenue
(années)
Angiomatose > 55 25
rétinienne
Hémangioblastome 35 - 55 29
cérébelleux
Cancer du rein 25 - 30 39 - 44
Kystes rénaux 25 44
Phéochromocytome 10 à 20 27
Hémangioblastome 10 à 15 30
médullaire
Kystes pancréatiques 10 à 15 37
Tableau 3. Comparaison des caractéristiques des tumeurs du
rein dans la maladie de Von Hippel-Lindau et dans les formes
sporadiques [15, 20, 23, 46, 55, 81].
Von Hippel-Lindau Forme sporadique
Age au diagnostic 39 ± 10 ans 61 ans
Bilatéralité 60 - 75% 1 - 2%
Multifocalité > 60% 7 - 20%

poser une attitude univoque qui doit prendre en comp-
te les caractéristiques propres de chaque patient (autres
localisations de la maladie de VHL, âge, acceptation de
l’hémodialyse...).
Indépendamment du cancer du rein, les autres manifes-
tations de la maladie de VHL nécessitent une sur-
veillance prolongée (Tableau 4).
Apparentés
• Calcul du risque:
Le dépistage génétique (*), disponible depuis peu, per-
met d’identifier les individus porteurs de l’anomalie
génétique, et de préciser individuellement le risque de
développer la maladie. Ce risque est de 70% à 60 ans
pour tout sujet ayant hérité de l’altération génétique
[56].Ce dépistage génétique est effectué individuelle-
ment par recherche directe de la mutation causale sur le
gène délétère à partir de l’ADN constitutionnel (*)
[97]. Cette technique permet d’identifier la mutation
dans environ 60% des cas [98]. Dans les autres cas, une
autre forme de dépistage génétique est possible, en
effectuant une étude familiale de liaison génétique (*).
Cette méthode, qui étudie la transmission dans la
famille de marqueurs chromosomiques (*) liés au gène
de prédisposition, nécessite cependant des fratries de
grande taille, et que plusieurs sujets atteints soient
vivants [35, 97].
• Dépistage clinique :
Si le dépistage génétique a pu être réalisé, le dépistage
clinique et morphologique ne s’adresse qu’aux sujets
porteurs de l’anomalie génétique [74, 98]. Dans le cas
contraire, il s’adresse à tous les membres de la famille
(Tableau 4).
Formes familiales de cancer du rein commun
Ces formes sont distinctes des précédentes car il
n’existe pas de syndrome particulier associé.
Il en existe deux types sur le plan histologique: les
adénocarcinomes non papillaires et les formes tubulo-
papillaires.
• Les tumeurs non papillaires (TNP) ont une agrégation
familiale(*) dans environ 1% des cas, alors que cette
forme histologique est la plus fréquente des cancers du
rein sporadiques (90% des cas): 25 familles compre-
nant 105 patients ont été recensées dans la revue de
MAHER en 1991 [73].
• Les tumeurs tubulopapillaires (TTP) sont des cancers
dont plus de 50% de la tumeur est composée d’un
contingent tubulopapillaire [119] et représentent 10%
des formes sporadiques [93, 119]. Elles sont générale-
ment hypo-vascularisées, et ont un bas grade de mali-
gnité dans 80 à 100% des cas [93] qui leur confère un
pronostic meilleur que les TNP [52, 93]. Cette forme
familiale a été peu rapportée dans la littérature puisque
dans une revue récente ZBAR et al. [120] n’ont recensé
que 40 patients issus de 9 familles (2 à 10 cas par
famille).
Aspects génétiques
Il persiste de nombreuses inconnues (Tableau 1).
Tumeurs non papillaires
• Mode de transmission
Il existerait une prédisposition génétique à transmis-
sion autosomique dominante, dont la pénétrance est
variable en fonction de l’âge [17, 36]. COHEN et al. ont
décrit, en 1979, une famille porteuse d’une transloca-
tion constitutionnelle t(3;8) (*) au sein de laquelle, sur
3 générations, 10 sujets présentaient un cancer du rein.
Le risque de développer un cancer du rein dans cette
famille était de 87% à 59 ans [17].
• Gènes impliqués
Il pourrait s’agir d’un gène suppresseur (*) situé en 3p
comme le gène VHL, mais sur un locus (*) différent
(3pl3-pl4 ?) [119].
346
Tableau 4. Maladie de Von Hippel-Lindau : modalités de surveillance des patients et des apparentés [97].
Lésion Dépistage Périodicité des examens
Début Patients Apparentés
Angiomatose rétinienne FO* 5 ans biannuel annuel
Hémangioblastome IRM 10 ans 1-3 ans 3-5 ans
sous-tentoriel
Hémangioblastome IRM 10 ans 1-3 ans 3-5 ans
médullaire
Cancer du rein Echo/TDM 15 ans biannuel annuel
Phéochromocytome Métanéphrines/TDM** 5 ans annuel annuel
Kystes pancréatiques Echo/TDM 15 ans annuel annuel
FO : fond d’oeil, IRM : imagerie par résonance magnétique, TDM : tomodensitométrie
* : angiographie si doute, ** : scintigraphie au M.I.B.G. en cas de suspicion.

Tumeurs tubulopapillaires
Le mode de transmission supposé est autosomique
dominant à pénétrance incomplète [119], et aucune
anomalie du chromosome 3 n’a été mise en évidence,
contrairement aux TNP [10, 52, 119].
Aspects cliniques
Ces tumeurs ont en commun l’âge de survenue précoce
(45 ans en moyenne), la bilatéralité, la multifocalité et
les récidives fréquentes [120]. C’est l’absence d’élé-
ment en faveur de la maladie de VHL (autres manifes-
tations de la maladie chez le patient ou dans sa famil-
le), qui permet le diagnostic de forme familiale de can-
cer du rein commun. Dans le cas particulier des formes
tubulopapillaires, la recherche d’une maladie de VHL
est inutile, car seuls des adénocarcinomes non tubulo-
papillaires sont associés à cette affection [97].
Implications pratiques
Patient
La notion de cancer du rein familial non papillaire
impose la recherche de maladie de VHL [97]. L’attitude
thérapeutique vis à vis du cancer du rein commun pose
les mêmes problèmes que pour la maladie de VHL
(bilatéralité, multifocalité, récidives). Pour les TTP, du
fait de lésions apparemment moins invasives, un traite-
ment chirurgical conservateur paraît moins risqué.
Apparentés
Les inconnues sur le mode de transmission, le (ou les)
gène(s) impliqué(s), et la rareté des anomalies obser-
vées en cytogénétique, conduisent à une surveillance
clinique de tous les membres de la famille. Dans le cas
particulier des TNP, il faut conseiller la pratique d’un
caryotype à la recherche d’anomalie du chromosome
3p (translocation, délétion) (*).
Tumeurs de Wilms familiales
Cette tumeur de l’enfant, d’origine embryonnaire est
associée dans 15% des cas à diverses malformations
[109]. Ces malformations, qui ne sont pas plus fré-
quentes dans les formes familiales [6] sont l’aniridie,
l’hémi-hypertrophie corporelle et des malformations
uro-génitales (cryptorchidie). Plus rarement, on obser-
ve certains syndromes malformatifs congénitaux, fami-
liaux ou non: syndrome W.A.G.R. (tumeur de Wilms,
aniridie, anomalie génito-urinaire, retard mental), syn-
drome de Beckwith-Wiedmann (S.B.W.), syndrome de
Denys-Drash, syndrome de Perlman [50, 108].
Fréquence
C’est la tumeur uro-génitale de l’enfant de moins de 15
ans la plus fréquente (80%), avec une incidence annuel-
le de 1 pour 10 000 enfants de moins de 16 ans. Les
formes familiales sont exceptionnelles (1% des cas) [6],
mais l’amélioration du pronostic devrait conduire dans
les années à venir à une augmentation de ces formes
familiales transmises par les patients survivants [78].
Aspects cliniques et histologiques
L’existence de caractéristiques cliniques et histolo-
giques propres aux TW familiales est discutée. Pour
certains auteurs les formes familiales de TW présentent
les caractéristiques des tumeurs à prédisposition géné-
tique par leur survenue plus précoce (âge moyen: 2,5
ans), et leur bilatéralité (20% des cas contre 3 à G%
dans les formes sporadiques [44, 50, 60, 78, 99].
Cette différence n’est cependant pas retrouvée dans
l’étude du N.W.T.S. (National Wilms Tumor Study) qui
comportait 65 cas familiaux [6].
Aspects génétiques (Tableau 1)
La maladie a une transmission autosomique dominan-
te, à pénétrance et expressivité (*) variables [78]. Il
existe 2 gènes de prédisposition, WT1(llpl3) et
5/WT2(11plS), qui sont également impliqués respecti-
vement dans le syndrome W.A.G.R. et S.B.W. La pro-
téine codée par le gène WT1 exerce son action comme
répresseur de la transcription, et interviendrait sur la
régulation de la croissance et de la différenciation du
rein et de l’appareil urogénital du foetus [22, 50]. Dans
certaines familles il n’existe pas de liaison aux loci pré-
cédents, ce qui témoigne de l’existence d’un gène
encore inconnu (WT3) [22, 42, 44, 101].
Implications pratiques
Patient
La recherche d’une lésion controlatérale et de malfor-
mations associées doit être systématique.
Un caryotype constitutionnel à la recherche de microdé-
létions sur le bras court du chromosome 11 est indiqué.
Sur le plan thérapeutique, le traitement des formes volon-
tiers multifocales et bilatérales (synchrones ou non) est
discuté. Le comité de chirurgiens du NWTS préconise,
en cas de tumeur bilatérale, un traitement conservateur se
résumant à des biopsies chirurgicales, suivies d’une chi-
miothérapie intensive (6 semaines à 6 mois) [54,109].
Une réintervention 6 mois plus tard (voire une 3ème à 1
an) peut s’accompagner d’une chirurgie partielle, avec
chimiothérapie et radiothérapie associées si l’exérèse
complète de la lésion est impossible.
D’autres auteurs ont cependant proposé une attitude
moins conservatrice [54]. Une surveillance prolongée
est nécessaire pour le diagnostic des récidives ou des
localisations controlatérales, ou pour détecter des com-
plications parfois tardives de la chimiothérapie.
Apparentés
Une consultation génétique peut être proposée même si
les formes familiales sont rares.
• En cas de tumeur de Wilms isolée, on recherche dans
347
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%