Nouveaux syndromes microdélétionnels

Dossier
Nouveaux syndromes
microdélétionnels
Alexandra Afenjar
1,2,3
, Lydie Burglen
1
1
Service de génétique, Hôpital Armand Trousseau, Groupement hospitalier universitaire Est,
26 avenue du Docteur Arnold-Netter, 75571 Paris Cedex 12
2
Service de neuropédiatrie, Hôpital Armand Trousseau, Groupement hospitalier universitaire
Est, 26 avenue du Docteur Arnold-Netter, 75571 Paris Cedex 12
3
Centre de référence anomalies du développement embryonnaire, Hôpital Armand
Trousseau, Groupement hospitalier universitaire Est, 26 avenue du Docteur Arnold-Netter,
75571 Paris Cedex 12
Résumé
Ces dernières années de nouvelles techniques permettant l’étude des chromo-
somes ont vu le jour. La fluorescence in situ hybridization ciblée sur les régions
télomériques, en particulier, a permis de mieux étudier ces régions terminales
des chromosomes. Ainsi des microdélétions terminales des chromosomes
inconnues auparavant ont été identifiées et de nouveaux syndromes cliniques
microdélétionnels ont pu être caractérisés. La description plus précise du
phénotype clinique, de l’éventuelle dysmorphie des problématiques médicales
et éducatives associés à ces nouveaux syndromes devraient permettre d’amé-
liorer la reconnaissance, la surveillance et la prise en charge des patients.
Mots clés : télomères, syndrome microdélétionnels
La mise au point des techniques
moléculaires d’étude des chromo-
somes a permis il y a une trentaine
d’années de mettre en évidence des
microdélétions à l’origine de syndro-
mes déjà bien connus phénotypique-
ment. Ainsi, on a pu démontrer que le
syndrome de Di George décrit en
1965 comme l’association d’une hy-
poplasie thymique, d’une hypocalcé-
mie par agénésie des parathyroïdes,
d’une dysmorphie faciale et d’une car-
diopathie de type conotroncal était lié
à une microdélétion 22q11. De
même, une microdélétion 7q11.2 a
été identifiée dans le syndrome de
Williams-Beuren décrit en 1961 et as-
sociant un retard psychomoteur avec
des difficultés d’apprentissage
contrastant avec un langage correct,
une cardiopathie (une sténose aorti-
que supravalvulaire le plus souvent),
une dysmorphie faciale et un phéno-
type comportemental caractéristique.
Plus récemment, les régions termi-
nales des chromosomes appelées télo-
mères ont été mieux étudiées. Elles
sont riches en gènes et contiennent
des séquences d’ADN répétitives plus
vulnérables, sièges plus fréquents de
réarrangements et de délétions. Ces
régions étant mal visualisées en cyto-
génétique, leurs remaniements ont
longtemps été méconnus. L’avène-
ment de nouvelles technologies telles
que la FISH (fluorescence in situ
hybridization) a permis d’identifier
des microdélétions terminales des
chromosomes inconnues auparavant
et ainsi de caractériser de nouveaux
m
t
p
Tirés à part : A. Afenjar
doi: 10.1684/mtp.2008.0171
mt pédiatrie, vol. 11, n° 4, juillet-août 2008
224
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

syndromes cliniques microdélétionnels. Les études réali-
sées dans des populations de patients avec retard mental
et caryotype normal ont montré que ces réarrangements
télomériques étaient une cause non rare des retards men-
taux inexpliqués chez l’enfant (de5à8%selon les études)
[1-3].
L’étude des télomères est un examen demandé de plus
en plus fréquemment lorsque les autres explorations (en
particulier le caryotype) n’ont pas permis d’établir un
diagnostic chez des patients avec retard mental ± syn-
drome malformatif et ± dysmorphie.
La description clinique précise de ces nouveaux syn-
dromes microdélétionnels devrait permettre une étude en
FISH plus ciblée en fonction de la symptomatologie ainsi
qu’une meilleure connaissance et donc une prise en
charge plus adaptée de ces pathologies.
Nous décrirons ici 3 syndromes : les délétions 22q13,
9q34 et 1p36.
Délétion 22q13
Le syndrome de délétion 22q13 résulte de la perte de
matériel génétique au niveau de la partie terminale du
bras long d’un des deux chromosomes 22. La délétion
22qter a tout d’abord été identifiée par la cytogénétique
conventionnelle (caryotype standard et haute résolution)
[4-6]. La FISH a ensuite permis de diagnostiquer de nou-
veaux patients porteurs de microdélétion non visible en
cytogénétique [7]. La sonde spécifique 22q13 étant utili-
sée comme sonde témoin de la sonde du syndrome vélo-
cardio-facial, certains diagnostics ont été faits de façon
fortuite lors de recherche de délétion 22q11 [8, 9].
La taille de la délétion est très variable, de 70 kb à prés
de 10 Mb et intéresse la bande terminale 22q13.3 [10].
Elle n’est visible sur le caryotype haute résolution que
dans moins d’un tiers des cas [11]. Elle est le plus souvent
isolée et de novo. Plus rarement, la délétion 22q13 est liée
à des translocations déséquilibrées, soit de novo, soit
résultant du déséquilibre de translocation héritée (10 %
des cas) [12]. La délétion peut aussi être associée à un
anneau du chromosome 22. Celui-ci est habituellement
en mosaïque. Enfin, au moins 2 patients sont rapportés
dans la littérature avec un syndrome de délétion 22q13 lié
à une délétion interstitielle épargnant le télomère [5, 13].
Récemment, le gène ProSAP2 ou SHANK3 a été iden-
tifié dans la région minimale critique du syndrome de
délétion 22qter [14]. SHANK3 code pour une protéine de
structure impliquée dans les synapses excitatrices. Il est
préférentiellement exprimé dans le cortex cérébral et le
cervelet [14]. La description d’un patient avec une trans-
location 12;22 interrompant SHANK3 et présentant un
phénotype similaire aux patients avec délétion 22qter a
conduit à impliquer l’haplo-insuffisance de ce gène dans
la genèse du phénotype neuropsychologique. Cette hypo-
thèse a ensuite été renforcée par l’identification de muta-
tions de SHANK3 chez des patients avec retard mental et
retard de langage sévère [15].
L’incidence de ce syndrome est inconnue à ce jour. Il
est également réparti dans les deux sexes.
Une large étude de Phelan et al. de 2001, sur 37 nou-
veaux patients et 24 de la littérature, a permis de préciser
le phénotype clinique. Il existe dans presque tous les cas
une hypotonie, un retard de développement psychomo-
teur et un retard mental.
L’hypotonie est précoce et peut disparaître complète-
ment avec l’âge. Le retard de développement psychomo-
teur peut être discret la première année avec une acquisi-
tion légèrement décalée de la tenue de tête et de la station
assise. Inversement, il peut être plus sévère avec absence
de tenue assise à 1 an. La marche acquise entre 15 mois et
3 ans est souvent ataxique.
Le retard mental est modéré à profond [12]. Les trou-
bles du langage parfois sévères peuvent être le signe initial
le plus évident (96 % des cas). Des traits autistiques sont
fréquemment associés et peuvent être au premier plan.
La croissance staturo-pondérale est normale ou, par-
fois, accélérée.
La dysmorphie est très discrète voire absente chez les
patients avec délétion cryptique infracytogénétique. Sont
décrits une dolichocéphalie (39 à 57 %), un ptôsis (13 à
57 %), un épicanthus (38 à 41 %), une racine du nez
épaisse et un nez proéminent, un petit menton pointu (11 à
62 %), des oreilles larges et/ou dysplasiques (47 à 65 %),
des mains et des pieds larges (25 à 68 %) et enfin une
syndactylie 2-3 des orteils (28 à 38 %) [10].
Une épilepsie est également rapportée dans près d’un
tiers des cas [8, 10-12].
D’autres symptômes plus occasionnels sont décrits :
malformations rénales, cardiaques, surdité, tolérance ac-
crue à la douleur, hypoplasie unguéale des pieds [12].
Lorsqu’il s’agit d’une translocation déséquilibrée, la
présence d’une trisomie partielle d’un autre chromosome
associée à la monosomie 22q13 peut modifier le phéno-
type. Mais la sévérité du phénotype clinique reste variable
même lorsque la délétion est isolée. Cette variabilité sem-
ble peu corrélée à la taille de la délétion [16].
L’hypotonie, le retard psychomoteur et le retard men-
tal sont des symptômes communs à de nombreux syndro-
mes, et la dysmorphie est discrète voire absente et peu
spécifique. L’atteinte sévère du langage, l’éventuelle
avance staturale et les traits autistiques sont plus caracté-
ristiques et doivent conduire à évoquer ce syndrome. De
plus, certains auteurs préconisent la recherche d’une mi-
crodélétion 22qter chez les patients autistes [8]. Lorsque le
caryotype haute résolution est normal, la délétion est
recherchée par FISH avec la sonde SHANK3. Si cette
recherche s’avère négative et qu’il existe une forte suspi-
cion clinique, un séquençage du gène SHANK3 pourrait
mt pédiatrie, vol. 11, n° 4, juillet-août 2008 225
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

se discuter mais n’est actuellement pas disponible en
diagnostic de routine.
Ces patients nécessitent une prise en charge en psy-
chomotricité et en kinésithérapie motrice afin de renforcer
le tonus musculaire. En l’absence de langage, la mise en
place de techniques de communication non verbale peut
être d’une grande aide.
Délétion 9q34
Le syndrome de délétion 9q34 est lié à la perte de
matériel génétique au niveau de la partie terminale du
bras long d’un des deux chromosomes 9.
Les deux premiers cas décrits ont été découverts de
façon fortuite lors de recherche systématique de réarrange-
ment télomérique chez des patients avec retard mental. La
plupart des patients rapportés ont une délétion non visible
sur le caryotype et seulement quelques cas visibles en
cytogénétique conventionnelle sont décrits. La taille de la
délétion est variable (700 kb à 2 Mb) [14] et les plus petites
délétions sont interstitielles et épargnent la partie toute
terminale du chromosome. La délétion est le plus souvent
pure et de novo, rarement dérivée de translocation.
Comme pour le syndrome de délétion 22qter, la des-
cription d’un patient présentant un phénotype compatible
et une translocation X;9 a fait suspecter un mécanisme
d’haploinsuffisance et permis d’identifier le gène respon-
sable du phénotype. Le gène interrompu par cette translo-
cation est le gène Eu-HMTase1 (euchromatic histone
methyl transferase) [18]. La responsabilité de ce gène dans
le phénotype des patients porteurs d’une délétion 9q34 a
été confirmée par l’identification de mutations chez des
patients avec un phénotype compatible mais sans délétion
[19]. Aucune différence significative n’a été mise en évi-
dence entre le phénotype des patients avec délétion et
celui de ceux porteurs d’une mutation du gène Eu-
HMTase1.
L’incidence de ce syndrome est inconnue à ce jour.
Il est également réparti dans les deux sexes.
Le phénotype est reconnaissable, associant dysmor-
phie faciale caractéristique, retard mental ± malforma-
tions viscérales [17, 20].
Le retard mental, l’hypotonie et le retard moteur sont
constants selon Stewart et al. dans une revue de la littéra-
ture portant sur 19 patients. Ils sont variables de modéré à
sévère. La marche est acquise entre l’âge de 18 mois et de
5 ans et un des patients ne marche pas à 11 ans. Le retard
de langage est très fréquent mais variable : certains
patients n’ont aucun langage, d’autres sont capables d’as-
socier des mots voire d’acquérir un niveau de langage
suffisant pour suivre une scolarité en milieu normal avec
aide. Rarement, des troubles du comportement de type
agressivité, polyphagie, troubles autistiques ou troubles
du sommeil sont observés.
Une épilepsie habituellement peu sévère et pharma-
cosensible est observée chez 35 % des patients [20].
La présence d’une cardiopathie de type conotroncale
est fréquente (50 %).
Des infections récurrentes des voies aériennes supé-
rieures et ORL sont décrites chez près de la moitié des
patients.
La dysmorphie faciale est caractéristique. Elle
comprend :
–un faciès grossier (50 %) plat avec hypoplasie de
l’étage moyen (65 %),
–un épicanthus (30 %) et un hypertélorisme (42 %),
–un synophris (70 %),
–un nez court avec des narines antéversées et une
racine large (75 à 85 %),
–une lèvre supérieure fine (12/14),
–une bouche en carpe avec lèvre inférieure éversée
(75 %),
–une macroglossie avec protrusion de la langue
(55 %),
–un micrognathisme (75 %).
La morphologie crânienne se caractérise par une bra-
chycéphalie (50 à 80 %). Il existe fréquemment une
microcéphalie vraie, anté- ou postnatale (PC ≤3DS) (65 à
85 %) ou un périmètre crânien dans les limites inférieures
de la normale.
La croissance staturo-pondérale est le plus souvent
normale mais l’installation d’un surpoids peut être obser-
vée (26 %) [20].
Diverses malformations occasionnelles ont également
été rapportées (rein, os, cerveau, tube digestif, organes
génitaux externes [hypospade, cryptorchidie]).
Chez ces patients un bilan malformatif doit être réalisé
afin de prendre en charge d’éventuelles malformations
viscérales. L’hypotonie et le retard psychomoteur nécessi-
tent une prise en charge en kinésithérapie motrice et
psychomotricité précoce. En raison du retard mental, une
aide doit être apportée pour la scolarisation de ces enfants.
Une orientation dans des structures d’éducation spéciale
est souvent nécessaire. Une prise en charge orthophoni-
que est nécessaire avec parfois la nécessité de mise en
place de technique de communication non verbale.
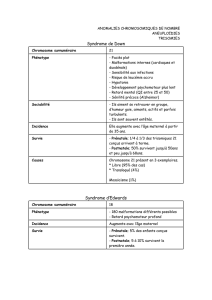
Délétion 1p36
Le syndrome de délétion 1p36 représente un des plus
fréquents syndromes microdélétionnels télomériques,
avec une prévalence estimée de 1/5 000 [21]. La perte de
matériel génétique au niveau de la partie terminale du
bras court d’un des deux chromosomes 6 est responsable
d’un syndrome reconnaissable appelé monosomie 1p36.
La délétion souvent non visible sur le caryotype est de
taille variable allant de 1,5 à 10,5 Mb. Elle est le plus
souvent pure et terminale mais peut également être
Nouveaux syndromes microdélétionnels
mt pédiatrie, vol. 11, n° 4, juillet-août 2008
226
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

interstitielle (7 %) ou résulter d’une translocation déséqui-
librée (15 %) [21]. Dans ce dernier cas, le phénotype peut
être influencé par la trisomie partielle d’un autre chromo-
some associé à la monosomie 1p36. Différents points de
cassure ont été décrits, ne permettant pas d’identifier une
zone minimale critique. Aucun gène n’a pu être mis en
cause à ce jour et la monosomie 1p36 est donc considérée
comme un syndrome de gènes contigus. Il n’y a pas de
corrélation génotype-phénotype bien établie.
Le syndrome, dominé par le phénotype neurologique,
associe un retard psychomoteur, un retard mental, une
hypotonie, une épilepsie, des troubles du comportement,
une microcéphalie, une dysmorphie et un retard de crois-
sance.
La dysmorphie reconnaissable est l’élément permet-
tant d’évoquer le diagnostic, associant une brachycépha-
lie, un front proéminent, des yeux enfoncés, des sourcils
horizontaux, des fentes palpébrales courtes, des coins des
lèvres tombants, un nez plat, des oreilles dysplasiques et
un menton pointu. La fontanelle est large et se ferme avec
retard. Une brachydactylie, une camptodactylie et des
pieds petits peuvent également être observés. Un épican-
thus, un palais ogival ou une fente labiopalatine sont
présents dans moins de 50 % des cas [22] (figures 1 et 2).
Le retard psychomoteur est souvent sévère, le retard
mental de modéré à profond et l’hypotonie marquée du-
rant la première année de vie. La marche est acquise chez
la plupart des malades mais reste instable avec une mau-
vaise coordination. Le développement du langage appa-
raît significativement altéré, soit absent, soit très pauvre
[22]. L’épilepsie, présente dans près de 60 % des cas,
débute pour la plupart des patients (70 %) dans les 6 pre-
miers mois de vie, rarement durant la période néonatale
avec des extrêmes de 1 mois à 7 ans [23]. L’EEG est
souvent précocement anormal. Différents types de crise
peuvent se voir : spasmes infantiles, crises généralisées
toniques, tonicocloniques ou cloniques. L’évolution de
l’épilepsie est généralement favorable mais selon les séries
10 à 50 % de pharmacorésistance est rapporté. L’IRM
cérébrale ne montre pas d’anomalies particulières. Quel-
ques patients avec un phénotype neurologique moins
sévère sont décrits. Ils n’ont pas d’épilepsie, un retard
mental léger et des capacités de communication verbales
satisfaisantes.
Il existe également chez ces patients avec monosomie
1p36 un phénotype comportemental particulier avec an-
xiété, agressivité, accès de colère et de violence avec
projection d’objets et automutilation (morsure des poi-
gnets) [23].
Des difficultés alimentaires sont fréquentes dans la
petite enfance, associées à un retard de croissance de
début pré- ou post-natal. Certains patients développent
par la suite une hyperphagie avec obésité responsable
d’un phénotype « Willi-Prader like » [24].
Des cardiopathies sont rapportées chez près de 50 %
des patients (canal artériel persistant, CIA, tétralogie de
Fallot, dysplasies valvulaires) [22]. Heilstedt et al. rappor-
tent 7 observations de cardiomyopathie dilatée durant la
première année de vie dans une étude portant sur 30 pa-
tients (23 %) [21]. Sur le plan endocrinien, une hypothy-
roïdie et une puberté précoce sont possibles.
On peut également observer une surdité et des anoma-
lies visuelles.
Il faut donc suspecter ce diagnostic devant une dys-
morphie évocatrice chez des patients avec retard psycho-
moteur, épilepsie, troubles du comportement et difficultés
d’alimentation précoces. Un phénotype « Prader-Willi »
(retard psychomoteur, hypotonie et difficultés d’alimenta-
Figure 1.Fille de 7 ans porteuse d’une délétion 1p36.
mt pédiatrie, vol. 11, n° 4, juillet-août 2008 227
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

tion initiaux puis polyphagie et obésité) non prouvé sur le
plan génétique doit faire discuter également ce diagnostic.
Ces patients nécessitent une prise en charge en psy-
chomotricité, kinésithérapie, orthophonie et en structures
éducatives adaptées. Il est essentiel de dépister et de traiter
une éventuelle épilepsie source de sur-handicap chez ces
enfants. Un retard dans la mise en route du traitement
et/ou un traitement inadapté en cas de spasmes infantiles
semble favoriser la survenue d’épilepsie réfractaire.
Il faut également prendre en charge les troubles du
comportement parfois invalidants.
Conclusion
Les nouvelles technologies ont permis d’identifier des
microdélétions des télomères. La description plus précise
du phénotype clinique et de l’éventuelle dysmorphie asso-
ciés à ces nouveaux syndromes microdélétionnels télomé-
riques permet de les évoquer et ainsi d’en faire la recher-
che par une FISH ciblée de la région suspectée, évitant
ainsi parfois une étude complète de tous les télomères plus
coûteuse. Des études de cohortes de patients ont com-
mencé à mieux préciser les problématiques médicales et
éducatives rencontrées et devraient ainsi améliorer la
surveillance et la prise en charge des patients.
Références
1. De Vries BBA, White SM, Knight SJL, et al. Clinical studies on
submicroscopic subtelomeric rearrangements : a checklist. J Med
Genet 2001 ; 38 : 145-50.
2. Flint J, Wilkie AO, Buckle VJ, et al. The detection of subtelomeric
chromosomal rearrangements in idiopathic mental retardation. Nat
Genet 1995;9:132-40.
3. Knight SJL, Regan R, Nicod A, et al. Subtle chromosomal rearran-
gements in children with unexplained mental retardation. Lancet
1999 ; 354 : 1676-81.
4. Herman GE, Greenberg F, Ledbetter DH. Multiple congenital
anomaly/mental retardation (MCA/MR) syndrome with Goldenhar
complex due to a terminal del (22q). Am J Med Genet 1988 ; 19 :
909-15.
5. Romain DR, Goldsmith J, Cairney H, et al. Partial monosomy for
chromosome 22 in a patient with del (22) (pter-q13.1 : :q13.33-qter).
J Med Genet 1990 ; 27 : 588-9.
6. Watt JL, Olson IA, Johnston AW, et al. A familial pericentric inver-
sion of chromosome 22 with a recombinant subject illustrating a
‘pure’ partial monosomy syndrome. J Med Genet 1985 ; 22 : 283-7.
7. Wong AC, Ning Y, Flint J, et al. Molecular characterization of a
130-kb terminal microdeletion at 22q in a child with mild mental
retardation. Am J Hum Genet 1997 ; 60 : 113-20.
8. Goizet C, Excoffier E, Taine L, et al. Case with autistic syndrome
and chromosome 22q13.3 deletion detected by FISH. Am J Med
Genet 2000 ; 96 : 839-44.
9. Precht KS, Lese CM, Spiro RP, et al. Two 22q telomere deletions
serendipitously detected by FISH. J Med Genet 1998 ; 35 : 939-42.
10. Phelan MC, Rogers RC, Saul RA, et al. 22q13 deletion syndrome.
Am J Med Genet 2001 ; 101 : 91-9.
11. Prasad C, Prasad AN, Chodirker BN, et al. Genetic evaluation of
pervasive developmental disorders : the terminal 22q13 deletion
syndrome may represent a recognizable phenotype. Clin Genet
2000 ; 57 : 103-9.
12. Havens JM, Visootsak J, Phelan MC, Graham Jr. JM. 22q13 dele-
tion syndrome : an update and review for the primary pediatrician.
Clin Pediatr (Phila) 2004 ; 43(1) : 43-53.
13. Fujita Y, Mochizuki D, Mori Y, et al. Girl with accelerated
growth, hearing loss, inner ear anomalies, delayed myelination of the
brain, and del (22)(q13.1q13.2). Am J Med Genet 2000 ; 92(3) :
195-9.
14. Bonaglia MC, Giorda R, Borgatti R, et al. Disruption of the Pro-
SAP2 gene in a t(12 ; 22)(q24.1 ;q13.3) is associated with the
22q13.3 deletion syndrome. Am J Hum Genet 2001 ; 69 : 261-8.
Figure 2.Fille de 5 ans porteuse d’une délétion 1p36.
Nouveaux syndromes microdélétionnels
mt pédiatrie, vol. 11, n° 4, juillet-août 2008
228
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
 6
6
1
/
6
100%