clonage d`un gène de meduse - ASSO-ETUD

OSINI Stéphanie, GALAN BRICEÑO Juana, MIRONOVA Elena, 14/11/2005
ORIGA Magali, BONVIN Grégoire Chimie/biochimie 2ème année
1
CLONAGE D’UN GÈNE DE MEDUSE
BIOCHIMIE DE L’ADN
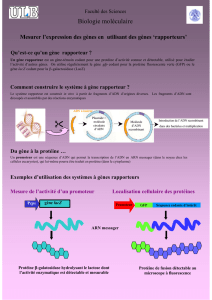
But de la manipulation
Le but de cette manipulation est l’introduction d’un gène de méduse
codant pour une protéine fluorescente dans des bactéries par l’intermédiaire
d’un vecteur (=plasmide dans notre cas précis), ce qui nous permettra de
cloner ce gène en prélude à une éventuelle purification de la protéine
d’intérêt.
Introduction
La GFD (green fluorescent protein) est une protéine naturellement
fluorescente trouvée dans des méduses (
Aequorea victoria
) vivant au fond de
la mer et leur servant à la communication par bioluminescence. Elle a été
isolée en 1962 par Shimomura et coll., et son gène a été cloné à partir de
1992 par Prasher et coll. C’est une protéine unique de 20kDa pouvant servir
de marqueur fluorescent, un puissant moyen d’études des protéines dans leur
contexte biologique.
En couplant une protéine d’intérêt par couplage avec une GFP, on peut par
exemple suivre sa localisation au sein de la cellule par microscopie de
fluorescence.
La fluorescence est produite par un réarrangement et l’oxydation de la
séquence Ser-Tyr-Gly, ce qui provoque la formation d’une double liaison et
une augmentation de la délocalisation électronique, provoquant l’émission de
lumière d’une longueur d’onde différente de celle absorbée (elle absorbe la
lumière UV pour émettre une lumière verte).
Un intérêt particulier de cette protéine est qu’elle peut être modifiée par
mutagenèse pour étendre ou réduire la délocalisation électronique, et donc de
modifier la couleur de fluorescence. On peut ainsi citer comme analogues à la
GFP :
BFP (bleu)
CFP (azur)
YFP (jaune)
…

OSINI Stéphanie, GALAN BRICEÑO Juana, MIRONOVA Elena, 14/11/2005
ORIGA Magali, BONVIN Grégoire Chimie/biochimie 2ème année
2
Image 1: Structure de la protéine GFP. Le réarrangement et l’oxydation de la
séquence Ser-Tyr-Gly est à l’origine de la fluorescence.
Pour une animation de la structure 3D de la GFP :
http://www.shef.ac.uk/~ifpm/gfpanim.html
Partie théorique
Dans le domaine de la biologie moléculaire, les chercheurs peuvent être
confrontés à un problème de ‘‘taille’’ : les protéines sur lesquelles ils
travaillent sont des molécules très petites à notre échelle et exercent le plus
souvent leur fonction dans le contexte de la cellule. Autant dire qu’il est
illusoire d’espérer pouvoir les voir avec un simple microscope.
Heureusement pour eux (et malheureusement pour nous, pauvres petits
étudiants, qui devons écrire un rapport sur le sujet) il existe une science
merveilleuse qu’est le génie génétique, permettant de produire une protéine

OSINI Stéphanie, GALAN BRICEÑO Juana, MIRONOVA Elena, 14/11/2005
ORIGA Magali, BONVIN Grégoire Chimie/biochimie 2ème année
3
en très grande quantité afin de mieux pouvoir l’étudier et la détecter, et
pouvant même permettre la localisation de la petite protéine dans l’immensité
de la cellule (et il ne reste plus qu’au chercheur à être capable de différencier
la membrane du réticulum endoplasmique de celle d’un lysosome qui passait
par là, mais c’est un autre problème).
Redevenons sérieux.
Aujourd’hui les chercheurs disposent d’outils puissants pour cloner un
gène en particulier et induire sa transcription en ARNm puis sa traduction en
protéine, incluant des moyen de localisation spatiale de la dite protéine et de
quantification.
Les premiers de ces outils sont les enzymes de restriction (aussi appelées
endonucléases de restriction), qui reconnaissent des séquences de bases
spécifiques dans l’ADN en double hélice et clivent les deux brins du duplex en
des points particuliers, tels des scalpels extraordinairement précis. Ces
enzymes sont utilisées dans de nombreux domaines, tels l’analyse de la
structure des chromosomes, le séquençage de très longues molécules d’ADN
ou l’isolement de gènes particuliers. Le chercheur suisse Arber a reçu un prix
Nobel en 1978 pour ces découvertes.
Les enzymes de restriction existent naturellement chez de nombreux
procaryotes où leur rôle est de cliver des molécules d’ADN étrangères, l’ADN
de la cellule n’étant pas dégradé car ne possédant pas la séquence
particulière reconnue par l’enzyme. Cette séquence, spécifique à chaque
enzyme de restriction, est dite
palindromique
: elle peut se lire de façon
identique de droite à gauche ou de gauche à droite, et la coupure de la
séquence peut se faire de deux manières :
à bout franc
(l’enzyme coupe
exactement au même endroit sur les deux brins) ou
cohésive
(l’enzyme coupe
en zigzag, ce qui laisse des extrémités monocaténaires).
Les enzymes de restrictions sont utilisées pour couper les molécules d’ADN
en fragments spécifiques au niveau de leur site de restriction, la séquence
qu’elles reconnaissent. De tels sites de restriction peuvent être introduits de
part et d’autre d’un gène d’intérêt afin de mieux pouvoir manipuler celui-ci.
Isoler un gène, c’est bien beau, mais encore faut-il ensuite trouver un
moyen de l’introduire dans une cellule pour que celle-ci puisse le transcrire.
On utilise pour ce faire un vecteur de clonage, qui doit être capable d’une
réplication autonome dans une cellule hôte donnée (qui doit donc posséder
son propre origine de réplication) et qui doit supporter l’insertion d’un
fragment d’ADN plus ou moins grand. Il existe plusieurs sortes de vecteurs de
clonages qui se différencient par leur conditions d’utilisation et la taille du
fragment d’ADN qu’ils permettent de cloner : les plasmides, les phages
lambda, les cosmides, les phages P1, les BAC (bacterial artificial chromosom)
et les YAC (yeast artificial chromosom).
Les plasmides permettent de cloner un fragment d’ADN jusqu’à 20kb. Il
s’agit de molécules d’ADN de petites tailles et circulaires, d’origine
bactérienne, pouvant être considérée comme un mini chromosome capable de
réplication autonome.

OSINI Stéphanie, GALAN BRICEÑO Juana, MIRONOVA Elena, 14/11/2005
ORIGA Magali, BONVIN Grégoire Chimie/biochimie 2ème année
4
Ainsi, un plasmide contient un ou plusieurs gènes de résistances à des
antibiotiques (permettant la sélection des cellules l’ayant intégré), une origine
de réplication et des sites de restriction permettant le clivage de son ADN
circulaire suivit de l’insertion d’un nouveau gène d’intérêt.
Avec un plasmide, on peut donc transformer une bactérie (typiquement
Escherichia coli
) pour qu’elle exprime le gène d’intérêt. Il suffit pour cela
d’intégrer le gène en question dans un plasmide par ouverture avec des
enzymes de restriction puis fermeture avec des enzymes de ligation, tel un
puzzle à l’échelle moléculaire, puis d’intégrer le plasmide dans la bactérie, qui
ainsi exprimera les gènes présents dans le plasmide, à commencer par la
résistances aux antibiotiques.
Arrivé à un tel stade, il convient de vérifier que le gène d’intérêt est bien
présent dans la bactérie. Sélectionner les bactéries ayant intégré le plasmide
n’est pas difficile, il suffit de les faire pousser sur un milieu contenant le ou les
antibiotiques contre lesquels les plasmides sont censés les protéger. Par
contre, vérifier la présence du gène d’intérêt ne peut se faire que par
migration sur gel d’électrophorèse à la suite d’une PCR.
La PCR (polymerase chain reaction) est une technique de réplication
élective
in vitro
d’ADN double brin, par extension itérative de deux amorces
appropriées, sous l’effet de l’alternance de températures variables :
Æ94°C pour la séparation (dénaturation) des brins matrices,
Æ30-65°C pour l’appariement des amorces, et
Æ72°C pour l’action de la
Taq
polymérase (polymérase thermostable
provenant d’une bactérie thermophile,
Thermus aquaticus
).
La répétition des cycles assure une duplication exponentielle de chaque
brin (le cycle dénaturation/appariement/synthèse étant répété de 20 à 40
fois, ce qui correspond à une amplification de 220 à 240). Par un choix
approprié des amorces, on peut n’amplifier qu’une portion bien définie de
l’ADN.
Les produits d’une PCR peuvent être analysés sur gel d’agarose, clonés ou
séquencés. La PCR est une technique puissante pour le diagnostic médical, la
médecine légale et l’évolution moléculaire, puisqu’elle permet de détecter, par
exemple, le virus du HIV-1 chez des sujets qui n’ont pas encore eu de
réponse immune à ce pathogène et chez lesquels le diagnostic par dosage
d’anticorps n’aurait donc pas pu être réalisé.
La bactérie a donc intégré le plasmide contenant notre gène d’intérêt,
dont la présence a été vérifiée par PCR et analyse sur gel d’agarose, et il ne
reste plus qu’à inciter la colonie ainsi sélectionnée (contenant des clones
identiques à la bactérie d’origine) à produire en masse la protéine
correspondante.
Pour cela, on utilise un promoteur particulier en amont du gène d’intérêt
qui s’active lorsqu’il reçoit un signal particulier, IPTG dans notre cas. La
bactérie se voit donc contrainte de transcrire et traduire encore et encore le
même gène, se remplissant de la protéine de méduse fluorescente, dont la
présence peut être aisément détectée, justement, par la fluorescence verte
qu’elle émet sous une lumière UV.

OSINI Stéphanie, GALAN BRICEÑO Juana, MIRONOVA Elena, 14/11/2005
ORIGA Magali, BONVIN Grégoire Chimie/biochimie 2ème année
5
Et pour récupérer la protéine en question, il suffit de détruire les bactéries
par ajout de produit la détruisant (TRIS, NaCl, lysosomes…) et d’ajouter des
billes de verres dans le lysat ainsi obtenu. Le gène de la protéine d’intérêt
ayant été associé à un court gène GST dont la protéine correspondante a la
particularité de se fixer sur les billes de verres, il ne nous reste plus qu’à
décoller GST-venus des billes de verres pour obtenir notre protéine purifiée,
fluorescente.
Merci les méduses.
Partie pratique
I)
Digestion du vecteur et de l’insert
L’insert (le gène GST-venus avec son promoteur IPTG et deux sites de
restriction spécifiques aux enzymes utilisées) et le plasmide (PGex4T-3
contenant le gène de résistance à l’ampicilline en plus des deux sites de
restrictions spécifiques aux enzymes utilisées) sont digérés séparément par
les enzymes de restriction BamHI et XhoI, en présence d’un tampon
spécifique, de BSA (rendant la viscosité optimale pour le bon fonctionnement
des enzymes) et H2O.
On fait migrer les deux solutions sur un gel d’agarose 1% afin de vérifier si
l’ADN est présent, c'est-à-dire qu’il n’y ait pas d’erreur de manipulation. Puis
les deux réactions sont incubées à 37°C toute la nuit.
Image 2 : dessins de l’insert et du plasmide avec les SR
II)
Purification de l’ADN
Les deux réactions sont purifiées par différents lavages avec du tampon
PB, qui fixe l’ADN, puis de la solution de lavage pour éliminer les enzymes
restantes et enfin H2O. Ces trois manipulations sont séparées par des
centrifugations. Cette étape de purification de l’ADN est indispensable afin
d’éliminer les enzymes de restriction qui seraient capables, sinon, d’attaquer
encore d’autres séquences.
Après la purification, on procède à la ligation afin de réunir le plasmide et
l’insert.
Deux tubes sont remplis avec le plasmide, le tampon de ligation et la ligase
(enzyme de ligation). Un des deux tubes contient également l’insert (ligation
n°2) tandis que l’autre tube sert de contrôle et l’insert est remplacé par de
l’eau (ligation n°1). Les ligations sont incubées à 16°C toute la nuit.
Image 3 : dessins du plasmide plus l’insert
III)
Transformation de la bactérie
Les bactéries sont tout d’abord traitées avec du CaCl2 ainsi qu’avec
diverses centrifugations, sous conservation dans la glace afin de les rendre
 6
7
8
6
7
8
1
/
8
100%