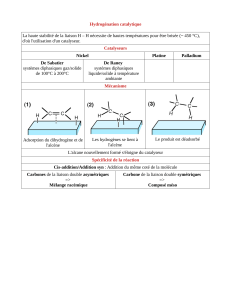
Chapitre III : Réactivité de la double liaison C=C I – Présentation de

L .PIETRI – Cours de Chimie Organique – Première année - Page 42 - 07/12/2008
Chapitre III : Réactivité de la double liaison C=C
I – Présentation de la double liaison
I-1) L’éthylène
I-2) Propriétés physiques
I-3) Réactivité
I-3-1 Définitions
I-3-2 Modes d’attaque
II – Addition électrophile ionique
II-1) Addition des dihalogènes : Halogènation
II-1-1 Présentation générale
II-1-2 Addition anti stéréospécifique
II-1-3 Mécanisme
II-1-4 Stéréochimie
II-2) Addition des halogènures d’hydrogène : Hydrohalogènation
II-2-1 Présentation générale
II-2-2 Mécanisme par carbocation
II-2-3 Règle de Markovnikov
II-3) Addition d’eau : hydratation
II-3-1) Présentation générale
II-3-2) Mécanisme réactionnel
II-3-3) régiosélectivité
III – Addition radicalaire
III-1) Présentation générale
III-2) Mécanisme
III-3) Stéréochimie
IV – Ozonolyse
IV-1) Formation d’un ozonide
IV-2) Réduction de l’ozonide
IV-3) Oxydation de l’ozonide
IV-4) Intérêt de l’ozonolyse

L .PIETRI – Cours de Chimie Organique – Première année - Page 43 - 07/12/2008
Chapitre III : Réactivité de la double liaison C=C
Introduction
La double liaison C=C se rencontre dans les alcènes, et plus généralement dans les dérivés
éthyléniques. Les alcènes sont peu fréquents à l’état naturel, mais de nombreux produits naturels
d’origine animale ou végétale, contiennent des molécules possédant une double liaison C=C.
On verra ainsi les additions de molécules sur la double liaison, puis on finira par l’ozonolyse…

L .PIETRI – Cours de Chimie Organique – Première année - Page 44 - 07/12/2008
I – Présentation de la double liaison
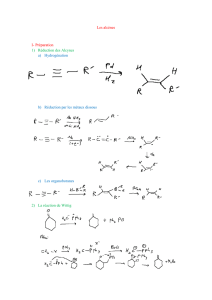
I-1) L’éthylène
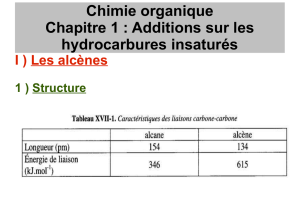
Les deux carbones fonctionnels trigonaux forment une double liaison : σ et π
• l
C=C
=134pm<l
C-C
=154
• D
C
=
C
=602 kJ.mol
-1
tel que 2D
C-C
>D
C=C
>D
C-C
L’énergie d’une liaison π est plus faible que celle d’une liaison σ. Les électrons π sont plus
mobiles et plus polarisables que les électrons σ.
I-2) Propriétés physiques
Dans les conditions usuelles, les termes légers (n<4) sont gazeux, les termes suivants liquides
volatils.
Les alcènes sont très peu solubles sont dans l’eau (mais plus que les alcanes en raison de
l’affinité du doublet π pour un solvant polaire)
Certains molécules possèdent un moment dipolaire, d’autres non (celui-ci demeure faible)

L .PIETRI – Cours de Chimie Organique – Première année - Page 45 - 07/12/2008
I-3) Réactivité
I-3-1 Définitions
Un réactif est une base de Lewis et un nucléophile lorsqu’il donne un doublet d’électrons pour
former une liaison avec un autre réactif.
Un réactif est un acide de Lewis et un électrophile lorsqu’il accepte un doublet d’électrons pour
former une liaison avec un autre réactif : ce dernier étant une base de Lewis ou électrophile.
Les termes qualificatifs de base et d’acide de Lewis concernent le caractère plus ou moins total
de la réaction correspondante. On considère l’aspect thermodynamique de la réaction.
Les termes qualificatifs de nucléophile ou électrophile concernent la vitesse avec laquelle la
liaison se forme. On considère l’aspect cinétique de la réaction.
I-3-2 Modes d’attaque
La double liaison C=C constitue un site à forte densité électronique : le doublet π de la liaison
C=C peut créer une liaison en apportant deux électrons.
• La liaison C=C est un site basique au sens de Lewis et nucléophile, il va donc subir des
attaques acides et électrophiles
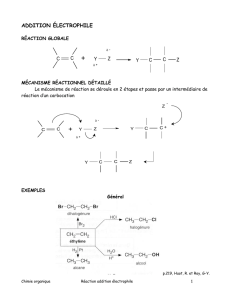
II – Addition électrophile ionique
II-1) Addition des dihalogènes : Halogènation
II-1-1 Présentation générale
• Bilan : R
2
C=CR
2
+ X
2
T R
2
CX-CXR
2
• Cinétique :
- la réaction de chloration est d’ordre global 2 : v = k[E].[Cl
2
]
- la réaction de bromation n’a pas d’ordre : v=k
1
[E].[Br
2
]+k
2
[E].[Br
2
]²+k
3
[E].[Br
-
][Br
2
]
• Influence des substituants :
- La vitesse augmente lorsque des atomes d’hydrogène liés aux atomes de carbone de
la double liaison sont remplacés par des groupes alkyles, elle diminue par
remplacement par un atome d’hydrogène.
• Influence du solvant : la vitesse augmente si la polarité du solvant augmente.
II-1-2 Addition anti stéréospécifique
• Une réaction chimique est stéréosélective si elle conduit préférentiellement ou exclusivement
à certains stéréoisomères parmi les possibles.
• Une réaction chimique est stéréospécifique si des stéréoisomères sont transformés
préférentiellement ou exclusivement en produits stéréochimiques différents.

L .PIETRI – Cours de Chimie Organique – Première année - Page 46 - 07/12/2008
Ainsi on peut dire que :
- L’addition de dihalogène est, en général, anti et stéréospécifique à 100%.
II-1-3 Mécanisme
a) Polarisation de Br
2
et attaque du nuage π
L’étape cinétique déterminante explique la loi de vitesse : v=k[E].[X
2
]
pour la chloration, le
mécanisme est plus complexe pour la bromation…
b) Attaque nucléophile anti.
Il y a attaque de l’ion ponté par l’ion bromure, à l’opposé du pont, car l’accès est plus facile.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%