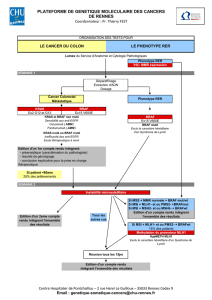
Traitement du mélanome par les inhibiteurs de BRAF

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
24
BRAF en oncologie
dossier thématique
Traitement du mélanome
par les inhibiteurs de BRAF
Melanoma treatment by BRAF inhibitors
Caroline Robert*, Christina Mateus*
* Service de dermatologie,
institut Gustave-Roussy,
Villejuif.
RÉSUMÉ
Summary
»Depuis la découverte de l’existence d’une mutation somatique
de l’oncogène BRAF dans 50 à 70 % des mélanomes, plusieurs
médicaments ciblant la sérine-thréonine kinase BRAF activée par
cette mutation ont été développés. Le chef de file est le vémurafénib
(Zelboraf®), inhibiteur puissant et spécifique de cette enzyme mutée.
Ce médicament entraîne des réponses objectives chez environ 50 %
des patients atteints de mélanome métastatique muté sur BRAF et
augmente significativement la durée de vie médiane par rapport
au traitement standard par chimiothérapie. Il vient d'obtenir une
autorisation de mise sur le marché en première ligne de traitement
dans cette indication. Les problèmes associés à cette thérapie sont
les échappements thérapeutiques fréquents et la survenue de
cancers cutanés secondaires illustrant des effets paradoxaux dans
certaines cellules de l'organisme. Des combinaisons de thérapies
ciblées sont prometteuses pour éviter ces 2 écueils.
Mots-clés : Mélanome – Inhibiteur de BRAF – Résistance –
Vémurafénib – Allongement de la survie globale.
Since the discovery of the existence of a somatic mutation
of BRAF oncogene in 50-70% of melanomas, several drugs
targeting the serine-threonine kinase BRAF activated
by this mutation have been developed. The leader is
vemurafenib (Zelboraf®), a potent and specific inhibitor of
this mutated enzyme. This drug induced objective responses
in approximately 50% of patients with metastatic melanoma
with mutated BRAF, and significantly increased median
survival when compared to standard treatment with
chemotherapy. It has obtained an authorization to market
in first line of treatment in this indication. The problems
associated with this therapy are the frequent secondary
resistances, and the occurrence of skin cancer which illustrates
the paradoxical effects of this drug in some cells. Combinations
of targeted therapies are promising to avoid both pitfalls.
Keywords: Melanoma – BRAF inhibitor – Resistance –
Vemurafenib – Lengthening of overall survival.
B
RAF était un gène assez peu connu jusqu’en
2002, avant la parution d’un article dans la
revue Nature qui nous a appris qu’il était
muté dans la majorité des lignées de mélanome (1).
Depuis, il est devenu un oncogène vedette, puisque
c’est celui qui est le plus souvent muté dans les tumeurs
humaines (environ 7 % toutes tumeurs confondues).
Rapidement après cette découverte majeure, il fut
confirmé que ce gène, codant pour une sérine-
thréonine kinase sur la voie des MAP-kinases, en aval
des protéines RAS et en amont de MEK (cf. article de
C. Longvert et L. Larue, p. 14), était muté dans environ
50 % des mélanomes. Dans la majorité des cas, il s’agit
d’une mutation récurrente V600E, qui entraîne une
activation de la protéine. Plusieurs données in vitro
suggéraient qu’un blocage de cette activation freinait
la prolifération tumorale, et il était donc logique de
développer des protocoles thérapeutiques ayant pour
objectif le blocage de cette kinase chez les patients
atteints de mélanome métastatique. Effectivement,
le traitement des patients souffrant de cette maladie
était jusqu’alors extrêmement décevant et reposait sur
des chimiothérapies successives qui n’avaient jamais
fait la preuve réelle de leur efficacité. Aucun médi-
cament n’avait été mis sur le marché depuis plus de
40 ans, et l’espérance de vie des patients était de moins
de 1 an en moyenne (2). Parallèlement, nous avions
assisté quelques années auparavant au développe-
ment rapide et efficace de l’imatinib, thérapie ciblée
capable de bloquer efficacement l’activité enzymatique
de la protéine de fusion BCR/ABL dans les leucémies
myéloïdes chroniques (LMC) et du récepteur KIT activé
par mutation ponctuelle dans les GIST. L’espoir est né
alors de pouvoir traiter les mélanomes sur le modèle
des GIST, avec des traitements ciblés efficaces et, si
possible, peu toxiques.

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
25
Traitement du mélanome par les inhibiteurs de BRAF
Développement des inhibiteurs de BRAF :
en tête, le vémurafénib
À l’époque, le développement du sorafénib dans le
cancer du rein était déjà bien avancé et on connaissait
son activité inhibitrice des protéines RAF, en plus de son
action antiangiogénique par inhibition des récepteurs
de VEGF et de PDGF. On espérait alors qu’il pourrait être
efficace sur les mélanomes du fait de son effet anti-RAF.
Cependant, le sorafénib fut un échec dans le traitement
du mélanome, car, après un résultat encourageant des
essais de phase I et II associant le sorafénib à une poly-
chimiothérapie par carboplatine et placlitaxel, 2 essais
de phase III conduits l’un en première ligne, l’autre chez
des patients prétraités, ne démontrèrent aucune supé-
riorité de l’association du sorafénib à cette polychimio-
thérapie par rapport à la chimiothérapie seule (3, 4).
C’est alors qu’apparut le PLX4032, que l’on connaît
maintenant sous la dénomination commune interna-
tionale de vémurafénib et sous le nom commercial de
Zelboraf®. Il s’agit d’un médicament bloquant plus spé-
cifiquement la protéine BRAF que les autres protéines
(ARAF et CRAF) ; son action est encore plus spécifique,
puisqu’elle s’exerce essentiellement sur la protéine BRAF
mutée en V600 (5). Il s’agit donc d’un inhibiteur puissant
et plus spécifique que le sorafénib. Les résultats de la
phase I furent publiés en 2009, ils étaient spectacu-
laires. Après quelques difficultés de formulation et une
fois que la dose efficace fut trouvée, il s’avéra que ce
médicament était efficace chez la majorité des patients
traités à partir du moment où leur mélanome était por-
teur de la mutation V600E (6). Cet essai démontra des
réponses objectives chez 56 % des patients, avec des
réponses cliniques et métaboliques visibles en TEP dès
les premières semaines de traitement.
La phase II fut conduite rapidement et porta sur
132 patients atteints de mélanome métastatique por-
teur de la mutation V600E ; elle confirma les résultats
très encourageants de la phase I avec 53 % de réponses
objectives, 29 % de patients stabilisés et une minorité de
patients ne répondant pas au traitement en première
intention (7). La durée de survie globale médiane des
patients inclus dans cet essai n’est pas encore connue
après 10 mois de suivi.
Enfin, une phase III à visée d’enregistrement a été
effectuée avant la fin de l’essai de phase II. Cet essai
multicentrique comparait le vémurafénib en première
ligne de traitement de patients atteints de mélanomes
de stade III ou IV non opérables à la chimiothérapie
standard par dacarbazine.
Cet essai fut conduit dans 12 pays et 104 centres ;
675 patients furent randomisés dans 2 bras en ouvert.
Les critères principaux de jugement étaient la survie
globale médiane et la survie sans progression. Le
recrutement s’effectua en 1 an, entre janvier 2010 et
janvier 2011. Quelques semaines après l’inclusion du
dernier patient, au vu des résultats obtenus, le comité
de sécurité de l’essai recommanda un amendement
autorisant un crossover. Cela autorisait les patients trai-
tés par dacarbazine et progressant sous traitement à
changer de bras de traitement et à recevoir le véruma-
fénib. Les résultats ont été publiés dans le New England
Journal of Medicine en juin 2011 (8). Les objectifs prin-
cipaux de l’essai ont été atteints, et la survie globale
est significativement augmentée par le vémurafénib
par rapport à la dacarbazine (HR : 0,44 ; p < 0,0001)
[figure 1]. On connaît maintenant, grâce aux données
actualisées en mars 2011, la survie médiane des patients
traités par dacarbazine ; elle est de 7,9 mois, comme on
pouvait s’y attendre. En revanche, on ne connaît pas
encore la survie médiane globale des patients traités
par vémurafénib, car plus de 50 % des patients sont
encore vivants. Les estimations actuelles évaluent à 83 %
le pourcentage de patients vivants après 6 mois dans le
bras vémurafénib, versus 63 % dans le bras dacarbazine.
En ce qui concerne le deuxième objectif de l’essai, la
survie sans progression, il est également atteint. La
survie sans progression médiane est de 1,6 mois pour
la dacarbazine et de 5,3 mois pour le vémurafénib. Le
taux de réponse objective est de 5,5 % versus 48,4 %
respectivement (figure 2).
Figure1. Courbe de Kaplan-Meier montrant la survie des patients au cours de l’essai BRIM 3.
Survie globale
(suivi médian avec vémurafénib : 6,2 mois)
Vémurafénib (n = 337)
Survie estimée à 6 mois = 83 %
Suivi médian = 6,2 mois
Dacarbazine* (n = 338)
Survie estimée à 6 mois = 63 %
Suivi médian = 4,5 mois
Hazard-ratio = 0,44
(IC
95
: 0,33-0,59) Dacarbazine
survie globale
médiane = 7,9 mois
100
90
80
70
60
50
40
30
20
10
0
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Temps (mois)
Survie globale (%)
Patients à risque (n)
Dacarbazine 338 302 268 232 186 145 111 83 43 26 12 5 0 0 0
Vémurafénib 337 336 334 317 285 218 178 125 82 54 22 11 4 2 0
Utilisation d’ipilimumab post-progression :
17 % de patients sous dacarbazine versus 6 % sous vémurafénib
* Les patients sous dacarbazine ayant reçu du vémurafénib après l’AI (selon la recommandation
du DSMB ; n = 50) ont été censurés à la date du crossover.

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
26
BRAF en oncologie
dossier thématique
Cette étude de phase III confirme que le vémurafénib
permet d’obtenir des réponses objectives chez environ
50 % des patients atteints de mélanome métastatique
et démontre qu’il est capable d’augmenter significa-
tivement la survie globale médiane de ces patients.
En ce qui concerne la toxicité du vémurafénib, peu
d’effets indésirables de grade 3 et 4 nécessitent l’arrêt
du traitement. La plupart des effets indésirables sont
de grade 1 ou 2. Les plus fréquents concernent les
articulations : il s’agit d’arthralgies d’allure inflam-
matoire chez près de 60 % des patients (tableau). On
observe aussi très fréquemment, chez plus de 30 %
des patients, des éruptions cutanées due à la photo-
sensibilité. Enfin, un effet indésirable particulière-
ment intriguant est la survenue, chez environ 20 %
des patients, de tumeurs cutanées d’origine kératino-
cytaire bénignes, intermédiaires, ou malignes à type
de papillomes cutanés, de kératoacanthomes ou
d’authentiques carcinomes épidermoïdes. Jusqu’à
présent, aucun de ces cancers cutanés ne s’est com-
pliqué de métastases, et ils sont facilement traités par
l’exérèse chirurgicale (figure 3).
Figure2. Courbe de survie sans progression au cours de l’essai BRIM 3.
Survie sans progression
(30 décembre 2010, analyse finale)
Hazard-ratio = 0,26
(IC95 : 0,20-0,33)
Test de log-rank, p < 0,0001
Vémurafénib (n = 275)
Dacarbazine
(n = 274)
Médiane = 5,3 mois
Médiane = 1,6 mois
100
90
80
70
60
50
40
30
20
10
0
0 1 2 3 4 5 6 7 8 9 10 11 12
Temps (mois)
Survie sans progression (%)
Patients suivis (n)
Dacarbazine 274 213 85 48 28 16 10 6 3 0
Vémurafénib 275 268 211 122 105 50 35 16 4 3
Tableau. Effets indésirables survenus au cours de l’essai BRIM 3.
Événements
indésirables (%)
Vémurafénib (n = 336) Dacarbazine (n = 287)
Tous Grade 3 Grade ≥ 4 Tous Grade 3 Grade ≥ 4
Arthralgie 53 4 – 3 < 1 –
Éruption cutanée 37 8 – 2 – –
Fatigue 38 2 – 33 2 < 1
Photosensibilité 33 3 – 4 – –
TFH 22 8 < 1 5* 1* –*
CCS cutané 17 16 – < 1 < 1 –
Kératocanthome 9 9 – – – –
Papillome cutané 21 < 1 – – – –
Nausée 35 2 – 43 2 –
Neutropénie < 1 – < 1 12 6 3
Uvéite** 3 < 1 – – – –
Arrêts dus à des effets indésirables** : 7 % pour le vémurafénib ; 4 % pour la dacarbazine.
* Données de l’AI de SG du 30 décembre 2010, non mises à jour pour le cutoff du 1er mars 2011. ** Données obtenues à partir d’un décompte manuel et non de résultats statistiques.
Figure3. Papillome cutané survenu quelques semaines
après le début d’un traitement par vémurafénib.

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
27
Traitement du mélanome par les inhibiteurs de BRAF
Le mécanisme d’apparition de ces cancers cutanés a
fait l’objet de plusieurs publications récentes. Ils pré-
sentent, dans un certain nombre de cas, des muta-
tions d’oncogènes tels que HRAS ou TP53 (9). On a pu
démontrer également que l’action d’un inhibiteur de
BRAF tel que le vémurafénib ou même le sorafénib était
capable d’entraîner, dans les cellules non porteuses de
la mutation de BRAF, une activation paradoxale de la
voie des MAP-kinases. Effectivement, comme cela avait
déjà été démontré dans des lignées de mélanomes non
mutés sur BRAF, en présence d’une activation de cette
voie par une mutation de NRAS par exemple, l’effet d’un
inhibiteur de BRAF conduit à une action inverse de ce
qui est observé dans les cellules porteuses de la muta-
tion avec une phosphorylation d’ERK, ce qui témoigne
d’une activation de la voie de signalisation (10, 11).
Nous avons démontré cet effet paradoxal sur les kéra-
tinocytes in vitro et dans des tumeurs et des peaux
normales de patients traités par sorafénib. Le modèle
que nous proposons est un modèle multi-étapes où
des mutations de certains oncogènes, potentiellement
induites par les effets des rayons ultraviolets sur la peau,
associées à l’effet de l’inhibiteur de BRAF et peut-être
à un effet combiné de certains virus présents sur la
peau comme les papillomavirus, pourraient conduire
dans certains cas à la transformation tumorale et à
l’apparition de kératoacanthomes et de carcinomes
épidermoïdes (12, 13). Il est intéressant de noter que
des mutations du récepteur 1 du TGFβ (TGFBR1) ont été
trouvées au sein de certaines tumeurs. Effectivement,
ce gène est connu pour être muté dans le syndrome
de Ferguson-Smith (syndrome des kératoacanthomes
multiples) [14]. Dans ce cas, il s’agit d’une mutation
germinale atteignant toutes les cellules de l’organisme,
alors que, dans les tumeurs kératinocytaires de patients
traités par sorafénib, les mutations sont somatiques et
ne sont retrouvées que dans les kératoacanthomes et
les carcinomes épidermoïdes.
Ces néoplasies épithéliales n’engagent pas le pronos-
tic vital des patients, mais, même si cet effet n’est pas
trop gênant – puisque les inhibiteurs de BRAF amé-
liorent par ailleurs leur survie –, il ne faut pas négliger
ce signal. Effectivement, la plupart des autres cellules
de l’organisme ne sont pas porteuses de la mutation
de BRAF, et, en théorie, il est possible de voir apparaître
au cours du temps d’autres proliférations néoplasiques.
D’ailleurs, tout récemment, 4 cas de mélanomes de
faible épaisseur ont été rapportés chez des patients
traités par vémurafénib (15).
À côté de l’apparition de ces tumeurs secondaires, un
autre écueil existe malheureusement lors de l’utilisation
du vémurafénib : il s’agit des résistances secondaires.
Effectivement, même si on observe environ 30 % de
réponses qui atteignent ou dépassent 12 mois, on
constate malheureusement que la majorité des patients
présentent une nouvelle progression tumorale après
6 à 8 mois de traitement.
Ces résistances secondaires font l’objet d’une intense
activité de recherche, et plusieurs mécanismes ont
déjà fait l’objet de publications. Dans certains cas, on
a retrouvé, dans les cellules résistant secondairement
au vémurafénib, des mutations de NRAS en amont
du blocage de BRAF ou encore une activation par
mutation de MEK, en aval de BRAF. On a également
trouvé une activation de MEK par activation d’une
protéine appelée COT. Dans d’autres cas, une autre
voie que la voie des MAP-kinases était activée, par
exemple par activation du récepteur de PDGF. Enfin,
plus récemment, des isoformes de BRAF, créées par
épissage alternatif, ont été identifiées comme cause
de cette résistance secondaire, puisque la protéine
ainsi créée, tronquée, n’est pas sensible au vémura-
fénib (10, 11, 16-19).
D’autres inhibiteurs de BRAF sont en cours de déve-
loppement. Le plus avancé après le vémurafénib est
le GSK2118436, développé par les laboratoires GSK,
qui a également donné des résultats très encou-
rageants en phase I et II. Les taux de réponse sont
similaires à ceux du vémurafénib. Le profil de toxicité
semble identique, si ce n’est l’absence de photosen-
sibilité, ce qui est clairement un avantage pour le
GSK2118436 (20).
D’autres inhibiteurs de BRAF, moins avancés dans leur
développement, sont également en cours d’évaluation.
Les perspectives
Les perspectives qui s’offrent à nous maintenant sont
multiples. Tout d’abord, la combinaison d’un anti-BRAF
avec un anti-MEK est une solution thérapeutique très
prometteuse. Cette combinaison est déjà évaluée en
phase I et II, et les résultats préliminaires, présentés à
l’ASCO en 2011, sont extrêmement encourageants (21).
Ce qui est très intéressant, c’est que certains patients
ayant résisté au vémurafénib sont parfois répondeurs
à cette association thérapeutique. Le deuxième avan-
tage majeur est que l’on n’observe quasiment pas de
kératoacanthome ni de carcinome épidermoïde avec
cette association. Plusieurs essais se mettent en place
pour en évaluer l’intérêt au stade métastatique, voire
même plus tôt, dans un contexte de traitement adju-
vant chez les patients de stade II ou III à haut risque de
récidive tumorale.

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
28
BRAF en oncologie
dossier thématique
L’épidémiologie moléculaire
pour accélérer la personnalisation
thérapeutique
D’après la 3e Journée de médecine translationnelle
et cancer du poumon
Coordonnateur: Jacques Cadranel (Paris) Sommaire
Éditoriaux
• Jacques Cadranel
• Gérard Zalcman
Les autorisations de mise sur le marché
conditionnées dans les cancers bronchiques
non à petites cellules aujourd’hui et demain
D’après la communication de D. Moro-Sibilot (Grenoble)
Quels biomarqueurs
pour quels traitements ?
• L’addiction oncogénique, une opportunité thérapeutique
• Résistances secondaires et thérapeutiques ciblées
• Personnaliser les traitements en dehors de l’addiction oncogénique
Comment développer
les biomarqueurs théranostiques ?
• De l’hypothèse à la validation
• De la validation à l’intégration dans un essai thérapeutique
• De l’essai thérapeutique à la diffusion en population générale
Comment accélérer
la personnalisation thérapeutique ?
• Projet Bio-France INCa/IFCT, un accès direct aux biothérapies
• Couple biomarqueur/thérapie ciblée,
un nouveau modèle de développement
• L’assurance qualité, une obligation sanitaire
Entretien avec Bernard Milleron
Entretien avec Frédérique Nowak
Supplément 1 au n° 3 vol. XXI
Mars 2012
Contact
Badia Mansouri : 01 46 67 62 74
Numéro réservé aux abonnés
Numéro réservé aux abonnés
Numéro réservé aux abonnés
VIENT
DE PARAÎTRE
D’autres stratégies thérapeutiques très séduisantes
sont des combinaisons de ces thérapies ciblées entre
elles – y compris celles inhibant la voie de signalisation
PI3-kinase, également impliquée dans l’oncogenèse du
mélanome (figure 4) – ou avec des immunothérapies. En
effet, il ne faut pas oublier qu’en parallèle des inhibiteurs
de BRAF, de MEK et d’autres protéines kinases, se déve-
loppe avec beaucoup de succès l’immunothérapie par
un anticorps anti-CTLA-4, l’ipilimumab (Yervoy®). Cette
immunothérapie non spécifique entraîne des réponses
chez environ 20 % des patients, indépendamment de
la présence d’une mutation de BRAF, et souvent très
prolongées. On envisage donc d’associer les thérapies
ciblées avec l’immunothérapie pour essayer d’obte-
nir des réponses non seulement fréquentes mais plus
durables et de combattre ainsi les résistances secon-
daires que l’on voit fréquemment apparaître sous
anti-BRAF. Un essai de phase I de combinaison entre
le vémurafénib et l’ipilimumab vient de commencer.
Conclusion
Des progrès majeurs ont été effectués ces dernières
années dans la compréhension de la biologie du méla-
nome et dans son traitement au stade métastatique. Les
inhibiteurs de BRAF, avec en tête de file le vémurafénib,
ont été développés très rapidement dans cette indica-
tion. Ils ont permis d’améliorer significativement le pro-
nostic de cette maladie et nous ont fait entrer dans l’ère
du traitement ciblé du mélanome. Des progrès restent
cependant à faire, et nous nous orientons progressive-
ment vers des stratégies de combinaisons thérapeu-
tiques afin de lutter contre les effets adverses et surtout
de contrer les résistances secondaires qui surviennent
fréquemment après quelques mois d’utilisation.
■
Conitd’intérêts.Caroline Robert est consultante pour Roche,
GSK et BMS.
Figure4. Schéma représentant les voies de signalisation des MAP-kinases et PI3-kinases. Les
différents types de mélanomes
(en haut)
comportent fréquemment des altérations d’une de
ces 2 voies. On s’oriente vers des stratégies de combinaisons de médicaments capables de bloquer
ces voies à différents niveaux.
NRAS
BRAF
50 % 15 %
MEK AKT 15 % 40 % deleted
Survie
Prolifération
PI3K PTEN
ERK mTOR
P16
CCND1
CDK4/6
2 %
1.
Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene
in human cancer. Nature 2002;417(6892):949-54.
2.Korn EL, Liu PY, Lee SJ et al. Meta-analysis of phase II coopera-
tive group trials in metastatic stage IV melanoma to determine
progression-free and overall survival benchmarks for future
phase II trials. J Clin Oncol 2008;26(4):527-34.
3.
Flaherty KT, Schiller J, Schuchter LM et al. A phase I trial of the
oral, multikinase inhibitor sorafenib in combination with car-
boplatin and paclitaxel. Clin Cancer Res 2008;14(15):4836-42.
4.Hauschild A, Agarwala SS, Trefzer U et al. Results of a
phase III, randomized, placebo-controlled study of sorafenib
in combination with carboplatin and paclitaxel as second-line
treatment in patients with unresectable stage III or stage IV
melanoma. J Clin Oncol 2009;27(17):2823-30.
5.Bollag G, Hirth P, Tsai J et al. Clinical efficacy of a RAF inhibitor
needs broad target blockade in BRAF-mutant melanoma.
Nature 2010;467(7315):596-9.
6.Flaherty KT, Puzanov I, Kim KB et al. Inhibition of muta-
ted, activated BRAF in metastatic melanoma. N Engl J Med
2010;363(9):809-19.
7.Ribas T, Kim K, Schuchter L et al. BRIM-2: An open-label,
multicenter phase II study of vemurafenib in previously trea-
ted patients with BRAF V600E mutation-positive metastatic
melanoma. J Clin Oncol 2011;29(suppl.):abstr. 8509.
8.
Chapman PB, Hauschild A, Robert C et al. Improved survival
with vemurafenib in melanoma with BRAF V600E mutation.
N Engl J Med 2011;364(26):2507-16.
9.Oberholzer PA, Kee D, Dziunycz P et al. RAS mutations are
associated with the development of cutaneous squamous cell
tumors in patients treated with RAF inhibitors. J Clin Oncol
2012;30(3):316-21.
10.Hatzivassiliou G, Song K, Yen I et al. RAF inhibitors prime
wild-type RAF to activate the MAPK pathway and enhance
growth. Nature 2010;464(7287):431-5.
11.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N.
RAF inhibitors transactivate RAF dimers and ERK signalling
in cells with wild-type BRAF. Nature 2010;464(7287):427-30.
12.
Arnault JP, Wechsler J, Escudier B et al. Keratoacanthomas
and squamous cell carcinomas in patients receiving sorafenib.
J Clin Oncol 2009;27(23):e59-61.
13.
Arnault JP, Mateus C, Escudier B et al. Skin tumors induced
by sorafenib; paradoxical RAS-RAF pathway activation and
oncogenic mutations of HRAS, TP53 and TGFBR1. Clin Cancer
Res 2012;18(1):263-72.
14.
Goudie DR, D’Alessandro M, Merriman B et al. Multiple self-
healing squamous epithelioma is caused by a disease-specific
spectrum of mutations in TGFBR1. Nat Genet 2011;43(4):365-9.
15.Dalle S, Poulalhon N, Thomas L. Vemurafenib in melanoma
with BRAF V600E mutation. N Engl J Med 2011;365(15):1448-9.
16.
Heidorn SJ, Milagre C, Whittaker S et al. Kinase-dead BRAF
and oncogenic RAS cooperate to drive tumor progression
through CRAF. Cell 2010;140(2):209-21.
17.Nazarian R, Shi H, Wang Q et al. Melanomas acquire resis-
tance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation.
Nature 2010;468(7326):973-7.
18.Poulikakos PI, Persaud Y, Janakiraman M et al. RAF inhibitor
resistance is mediated by dimerization of aberrantly spliced
BRAF(V600E). Nature 2011;480(7377):387-90.
19.
Wagle N, Emery C, Berger MF et al. Dissecting therapeutic
resistance to RAF inhibition in melanoma by tumor genomic
profiling. J Clin Oncol 2011;29(22):3085-96.
20.Keeford R, Long G, Arkenau HT et al. Selective inhibition of
oncogenic BRAF V600E/K/D by GSK2118436: Evidence of clinical
activity in subjects with metastatic melanoma. Pigment Cell
Melanoma Res 2010;23:912.
21.
Infante J. Phase I/II study to assess safety, pharmacoki-
netics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212
(GSK212) dosed in combination with the oral BRAF inhibitor
GSK2118436 (GSK436). J Clin Oncol 2011;29(suppl.):abstr.
CRA8503.
Références
1
/
5
100%