Métabolisme du fer et cancer du sein : connaissances et perspectives

doi:10.1684/abc.2012.0700
387
Pour citer cet article : Durigova A, Jacot W, Pouderoux S, Roques S, Montels F, Lamy PJ. Métabolisme du fer et cancer du sein : connaissances et perspectives. Ann Biol
Clin 2012 ; 70(4) : 387-96 doi:10.1684/abc.2012.0700
Synthèse
Ann Biol Clin 2012 ; 70 (4) : 387-96
Métabolisme du fer et cancer du sein :
connaissances et perspectives
Iron metabolism in breast cancer: knowledge and future
Anna Durigova1
William Jacot1
Stéphane Pouderoux1
Sylvie Roques2
Frédéric Montels2
Pierre-Jean Lamy2
1Service d’oncologie médicale,
2Laboratoire de biologie spécialisée
et d’oncogénétique, CRLC Val
d’Aurelle-Paul Lamarque, Montpellier
<pierre-
jean.lamy@montpellier.unicancer.fr>
Article rec¸u le 13 décembre 2011,
accept´
e le 20 février 2012
Résumé. Le fer joue un rôle fondamental en biologie et sa concentration dans
les organismes vivants est régulée de fac¸on très précise. De nombreuses molé-
cules de stockage et de transport sont utilisées pour maintenir cette homéostasie
intracellulaire. Les cellules cancéreuses présentent des altérations de cet équi-
libre, et la présence d’un cancer induit de profondes dysfonctions dans le
métabolisme martial. Des études récentes ont montré que les cellules cancé-
reuses mammaires étaient affectées d’anomalies impliquant plusieurs protéines
comme la ferroportine et l’hepcidine. Un impact pronostique de ces perturba-
tions a été rapporté chez des patientes atteintes d’un cancer du sein et certaines
molécules régulatrices du métabolisme martial pourraient devenir des cibles
thérapeutiques. Il s’agit d’une approche innovante qui se dessine pour traiter un
cancer qui reste, malgré les progrès des traitements et l’apparition des thérapies
ciblées, la première cause de mortalité par cancer chez la femme.
Mots clés : fer, cancer du sein, ferritine, ferroportine, hepcidine
Abstract. Iron plays a fundamental role in biology and its concentration in
living organisms is regulated very precisely. Many molecules of storage and
transportation are used to maintain the intracellular homeostasis. Cancer cells
have alterations in this balance. Recent studies have shown that breast cancer
cells present abnormal expression of several proteins such as hepcidin and fer-
roportin. A prognostic impact of these alterations has been reported in patients
with breast cancer. Regulatory molecules of iron metabolism could become the-
rapeutic targets. This is an innovative approach that has emerged for treating
a cancer which, despite advances in treatment and the emergence of targeted
therapies, remains the leading cause of cancer death in women.
Key words: iron, breast cancer, ferritin, ferroportin, hepcidin
Le fer est le sixième élément le plus abondant dans
l’univers. Il est fondamental et indispensable pour la biolo-
gie des cellules eucaryotes. Retrouvé dans de nombreuses
hémoprotéines comme l’hémoglobine ou la myoglobine,
il participe également à la respiration cellulaire dans
les cytochromes ainsi qu’aux réactions d’oxydoréduction
catalysées par des enzymes ferro-dépendantes, les ribo-
nucléotides réductases et les xanthine-oxydases, qui
interviennent au niveau de l’ADN. Il intervient encore
sur les cyclo-oxygénases et les lipo-oxygénases de
l’inflammation. Le fer fonctionnel est impliqué dans la
fonction des catalases et des peroxydases qui protègent
de la formation des radicaux libres. La régulation de son
taux doit être fine, car un excès de fer libre est rapide-
ment toxique et une carence induit une hypoxie par anémie.
La connaissance de son métabolisme chez l’homme s’est
considérablement élargie ces dernières années avec la
découverte de nombreuses molécules impliquées dans
le stockage, le transport dans la circulation sanguine et
les mécanismes de régulation des concentrations intra et
extracellulaires. De nombreuses études ont décrit les ano-
malies du métabolisme du fer que présentent les cellules
tumorales et les patients atteints de cancer. Plus récemment,
Tirés à part : P.-J. Lamy
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

388 Ann Biol Clin, vol. 70, n◦4, juillet-août 2012
Synthèse
le rôle de ces anomalies a été confirmé dans des études
fondamentales et cliniques en cancérologie mammaire [1].
Cette revue se propose de faire le point sur ce sujet en résu-
mant nos connaissances actuelles sur le métabolisme du
fer et les anomalies de ce dernier dans les cancers lors des
anémies dites inflammatoires. Puis, nous détaillerons spéci-
fiquement les anomalies martiales décrites dans les cancers
du sein, la valeur pronostique des nouveaux marqueurs,
ainsi que les cibles thérapeutiques potentielles visant le
métabolisme du fer.
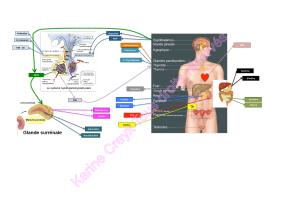
Le métabolisme du fer
et les marqueurs biologiques
du bilan martial
Le métabolisme du fer
L’organisme contient3à5gdeferquiestquasiment tota-
lement recyclé au cours de son métabolisme. Seuls 2 mg de
fer sont absorbés chaque jour en compensation des pertes
urinaires, cutanées et digestives. Dans les conditions nor-
males, la plus grande partie du fer est étroitement liée à des
molécules fonctionnelles comme l’hémoglobine (2,6 g) ou
la myoglobine (0,4 g), à des protéines de transport comme
la transferrine (3 mg) ou à des protéines de stockage intra-
cellulaire comme la ferritine (1 g de fer est stocké dans le
foie). Dans cet état fortement lié, le fer ne peut pas causer
de lésions cellulaires, car il est incapable de participer à des
réactions d’oxydation.
L’absorption du fer se fait essentiellement sous forme hémi-
nique. Les protéines héminiques sont digérées : l’hème est
libéré et absorbé par les entérocytes grâce à un transpor-
teur de l’hème, la protéine HCP (heme carrier protein)
découvert en 2005. Le fer non-héminique est d’abord réduit
par l’acidité gastrique et son absorption par l’entérocyte est
influencée par la présence des autres nutriments. Le passage
du fer non-héminique sous forme de fer ferreux Fe2+ vers
le cytoplasme de l’entérocyte au niveau de la membrane
apicale est assuré par le dimetal transporter 1 (DMT1)
qui assure également la réduction du fer. La capacité
d’absorption du fer au niveau des entérocytes est modulée
par les signaux provenant des autres cellules de l’organisme
qui interviennent dans la consommation (précurseurs éry-
throïdes) et le stockage (hépatocytes, macrophages) du
fer. La diminution de l’expression de la protéine DMT1
au niveau de l’entérocyte, en cas de réserves impor-
tantes en fer, expliquerait la diminution de son absorption
[2].
L’absorption du fer est augmentée en cas de carence
martiale ou en cas d’augmentation de l’érythropoïèse,
et à l’inverse diminuée en cas de surcharge martiale.
Cette régulation, très complexe, fait aussi intervenir des
signaux inflammatoires et hypoxiques, notamment par
l’intermédiaire de l’hepcidine [3].
Avant son excrétion par l’entérocyte, le Fe2+ cytoplasmique
est oxydé en Fe3+ par une ferroxidase, l’hephaestine. Le
Fe3+ intracellulaire est ensuite excrété par la ferroportine au
niveau du pôle latéro-basal de l’entérocyte. La ferroportine
permet sa fixation à la transferrine (encore appelée sidéro-
philine), son transporteur plasmatique qui assure l’apport
de Fe3+ aux érythroblastes. La ferroportine est également
appelée iron regulated transporter 1 ou SLC40A1 [3]. Elle
est indispensable à l’exportation du fer de l’entérocyte vers
le plasma. Ainsi les souris homozygotes présentant une
délétion du gène de la ferroportine ne peuvent pas excréter
le fer et présentent une accumulation massive du fer dans
les entérocytes. Enfin, le fer qui n’est pas transféré vers le
plasma est éliminé, avec les entérocytes dont la durée de
vie est très courte (en moyenne de sept jours dans le duo-
dénum et le jéjunum et de trois jours dans l’iléon), dans les
matières fécales.
La moelle osseuse est le premier consommateur de fer. Le
fer entre dans les précurseurs érythroïdes grâce aux récep-
teurs à la transferrine de type 1 (TfR), qui, une fois la
transferrine fixée, s’internalisent sous forme d’une vacuole
d’endocytose. Le pH acide dans cet environnement permet
la libération du Fe3+ de la transferrine. Une fois libéré, le
Fe3+ excrété est non réactif et soluble. Le Fe3+ est réduit par
la cytochrome b réductase membranaire et son internalisa-
tion dans le cytoplasme se fait par le transporteur DMT1. Il
forme alors un pool labile de fer fixé à différentes protéines
ferro-dépendantes, avec un rôle particulièrement important
au niveau de la mitochondrie. Toute surcharge en fer dans la
cellule est prise en charge par la ferritine, une molécule de
stockage. L’exportation du fer des cellules de l’organisme
vers la circulation sanguine utilise le même mécanisme avec
pour ferroxidase la céruléoplasmine [4].
L’homéostasie du fer se fait uniquement au niveau de
l’absorption et du recyclage du fer par les macrophages.
Le contrôle majeur de la sidérémie se fait par le biais de
l’hepcidine, une protéine synthétisée par le foie [5]. Son
rôle est de diminuer l’absorption intestinale du fer. Elle
augmente dans le même temps la rétention intracellulaire
du fer par les macrophages et les hépatocytes. Une aug-
mentation de sa production se retrouve dans les tumeurs
à hepcidine [6], induisant une anémie ferriprive sévère
et dans les anémies inflammatoires en réponse à la sur-
charge en fer, entraînant une séquestration de celui-ci. Le
rôle de l’hepcidine sur l’homéostasie du fer a été éta-
bli sur les modèles de souris transgéniques. L’injection
d’hepcidine synthétique provoque chez la souris une hypo-
sidérémie dans l’heure qui suit son administration [7]. Une
déficience complète en hepcidine conduit à une hémochro-
matose juvénile, forme sévère de surcharge martiale dans
laquelle l’absorption intestinale du fer est altérée, le fer
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Ann Biol Clin, vol. 70, n◦4, juillet-août 2012 389
Fer et cancer du sein
continuant à être absorbé malgré les stocks excessifs. À
l’inverse, la production excessive de l’hepcidine conduit à
la carence martiale et à l’anémie en raison de l’incapacité
à absorber le fer au niveau intestinal malgré des apports
en fer normaux ou augmentés. La sévérité de ces deux états
pathologiques fait supposer qu’il n’existe probablement pas
de mécanisme compensateur des effets de l’hepcidine. Le
rôle de l’hepcidine dans l’homéostasie du fer semble donc
crucial [3].
L’hepcidine agit en se fixant sur une boucle extra-
membranaire de la ferroportine, conduisant à la phos-
phorylation de cette protéine et à son internalisation puis
sa destruction dans l’entérocyte par le protéasome [8].
Elle est elle-même régulée par des facteurs inhibiteurs
comme l’augmentation des besoins de l’érythropoïèse ou
l’hypoxie, et des facteurs stimulant son expression comme
l’augmentation de la sidérémie et l’inflammation. En cas
d’inflammation, l’expression d’hepcidine est induite par les
lipopolysaccharides (LPS) et l’interleukine 6 (IL-6). L’IL-
6 active la synthèse de l’hepcidine par la voie du facteur
de transcription STAT3 [9]. Ce mécanisme permet de faire
le lien entre la réponse immunitaire, l’homéostasie du fer
et l’anémie inflammatoire associée au cancer. La produc-
tion de l’hepcidine est inhibée en cas d’augmentation de
l’érythropoïèse [10].
Initialement découverte pour son rôle antimicrobien, il
semble que cette molécule soit issue de l’évolution de
systèmes de défense de l’organisme, diminuant le fer
disponible pour les agents pathogènes ou les cellules tumo-
rales. De nombreuses autres protéines sont impliquées
dans la régulation de l’absorption du fer comme la pro-
téine HFE (Human hemochromatosis protein) codée par
le gène HFE hemochromatosis dont les mutations sont
responsables de la principale forme d’hémochromatose
primitive. Dans ce cas, les patients sont atteints d’un
déficit en hepcidine que l’on retrouve dans d’autres
formes d’hémochromatose héréditaire [11]. Dans le cas
de l’hémochromatose juvénile de type 2AB, la surcharge
en fer est liée directement à une mutation du gène de
l’hepcidine (HAMP, hepcidin antimicrobial peptide) sur le
chromosome 19.
La régulation du métabolisme du fer intracellulaire au
niveau des cellules non érythroblastiques est basée sur le
contrôle de la synthèse de la ferritine et du TfR, au stade
post-transcriptionnel par des iron regulatory proteins dont
l’action est modulée directement par la concentration en fer
dans le pool labile, le fer non fixé à la ferritine et directement
mobilisable [4].
Les marqueurs biologiques du bilan martial
L’exploration du métabolisme du fer repose sur un nombre
restreint d’examens [12]. Elle peut se réaliser à l’aide de
la cinétique d’absorption de l’isotope 59Fe, rarement prati-
quée. Le taux de fer circulant est un paramètre qui subit des
variations nycthémérales importantes avec des maximas à
midi et des minimas le soir. Son exploration est basée sur
le dosage du fer plasmatique prélevé à jeun le matin et cou-
plé au dosage de la transferrine. Les valeurs usuelles sont
(pour des prélèvements effectués le matinà8h):homme :
9à30mol/L, femme:8à28mol/L. La transferrine
est une bêtaglobuline capable de fixer 2 ions Fe3+. On peut
ainsi calculer le coefficient de saturation de la transferrine
(CST), selon la formule : CST (%) = [concentration en fer
plasmatique (mol/L)] / [25 x concentration en transferrine
(g/L)] x 100. La valeur usuelle du CST est comprise entre
20 et 40 %. Autre marqueur du bilan martial, la ferritine est
une protéine capable de fixer plus de 4 000 atomes de fer
qui permet d’explorer les réserves en fer de l’organisme,
soit 15 à 30 % du fer total. Sa valeur normale est de 50
à 350 ng/mL chez l’homme et de 30 à 120 ng/mL chez
la femme. Le TfR est une protéine composée de deux
sous-unités capable de fixer la transferrine [13]. Elle peut
être clivée pour donner le TfRs, fragment sérique dont
la concentration reflète l’expression cellulaire du récep-
teur. Comme la numération des réticulocytes, le TfRs
est un marqueur sanguin du fer fonctionnel qui mesure
l’efficacité de l’érythropoïèse en évitant d’avoir recours
à des biopsies de moelle osseuse, la diminution du fer
fonctionnel s’accompagnant d’une augmentation du TfRs
[14]. Une revue de la littérature concernant l’utilisation du
TfRs en tant que marqueur du statut martial a été publiée
en 2009 [15], 9 essais ont été retenus, les auteurs ont
conclu que le dosage plasmatique de TfRs peut améliorer
le diagnostic de l’anémie dans les situations complexes où
l’anémie inflammatoire et la carence martiale coexistent.
Le TfRs est augmenté dans la carence martiale (où le fer
n’est pas disponible pour l’érythropoïèse), alors que dans
l’anémie inflammatoire le TfRs est normal car négative-
ment régulé par les cytokines inflammatoires. En dehors
des pathologies liées à une stimulation de l’érythropoïèse,
une augmentation du TfRs est toujours associée à un déficit
en fer.
Devant la complexité du diagnostic d’une carence mar-
tiale en cas de syndrome inflammatoire associé, d’autres
marqueurs ont été recherchés. Le contenu en hémoglo-
bine des réticulocytes (CHr) est un paramètre qui permet
d’estimer la disponibilité du fer de la moelle osseuse pour
l’érythropoïèse dans les 2 jours qui ont précédé le dosage
et donc permet de dépister les états de carence martiale
relative, de manière extrêmement précoce. Le pourcentage
d’hématies hypochromes représente directement la manière
dont le fer est utilisé dans la synthèse de globules rouges.
La ferritine érythrocytaire est un résidu de la ferritine éry-
throblastique. Elle est le reflet de la balance entre le fer
disponible dans la moelle osseuse et le fer utilisable pour
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

390 Ann Biol Clin, vol. 70, n◦4, juillet-août 2012
Synthèse
la synthèse de l’hémoglobine. La baisse de la ferritine
érythrocytaire témoigne d’une carence martiale vraie même
si une anémie inflammatoire coexiste [16].
Enfin, compte tenu du rôle majeur du fer dans la matu-
ration des érythrocytes, les paramètres hématologiques
(hémoglobinémie, hématocrite, nombre d’érythrocytes,
concentration hémoglobinique moyenne ou CCHM et
volume globulaire moyen ou VGM) constituent des
marqueurs indirects, peu sensibles et peu spécifiques,
d’évaluation du fer fonctionnel.
Les anomalies du bilan martial dans
les cancers
Les perturbations, au moins indirectes, du métabolisme du
fer dans les pathologies cancéreuses pouvaient être suspec-
tées depuis longtemps. En effet, l’anémie inflammatoire est
un tableau fréquemment retrouvé au cours du cancer [17].
Or, la pathologie cancéreuse est fréquemment associée à
un tableau inflammatoire chronique. Cet état inflammatoire
peut conduire à un type d’anémie initialement normo-
chrome, normocytaire, arégénérative qui se traduit par une
baisse du CST liée à une diminution du fer plasmatique
[18]. La cause n’est pas une carence d’apport en fer puisque
les réserves de fer ne sont pas abaissées. En réalité, cette
hyposidérémie est le résultat d’un stimulus inflammatoire
très puissant lié à la sécrétion importante de cytokines pro-
inflammatoires (IL-1a, IL-6, interféron gamma, TGF bêta,
TNF alpha) par les cellules tumorales et les macrophages
infiltrant le tissu tumoral. La lignée érythrocytaire est par-
ticulièrement affectée par cette inflammation chronique.
La différenciation des progéniteurs érythrocytaires médul-
laires est inhibée par ces cytokines. Elles inhibent aussi la
synthèse de l’érythropoïétine (EPO) et entraînent des phé-
nomènes de résistance à l’EPO [19]. Enfin, elles agissent
directement sur le métabolisme du fer en induisant une
séquestration martiale, expliquant la possible évolution de
ces anémies vers l’hypochromie et la microcytose (signes
toutefois moins marqués que dans les anémies par carence
martiale vraie).
En effet, l’interféron gamma et le TNF␣induisent
l’expression de DMT1 et du TfR ayant pour conséquence
une augmentation de la captation du fer par les macro-
phages qui, par ailleurs, augmenterait dans le même temps
leurs propriétés antitumorales. Les interleukines 1 et 6 ainsi
que le TNF␣stimulent la production de ferritine. Aussi,
l’augmentation de la ferritine plasmatique est une anoma-
lie fréquemment observée au cours des cancers. Lors de
la réponse immunitaire, l’IL-6 et les LPS stimulent la pro-
duction hépatique de l’hepcidine qui inhibe l’absorption
duodénale du fer et favorise la séquestration macrophagique
du fer tout en limitant son recyclage. Ces mêmes stimuli
inflammatoires diminuent l’expression de la ferroportine
au niveau des entérocytes duodénaux, par conséquent le
transport du fer de l’entérocyte vers la transferrine est
bloqué. Un autre mécanisme est lié à l’interleukine 10
(IL-10). Cette dernière augmente l’expression des récep-
teurs de la transferrine sur les macrophages et améliore
leurs capacités d’importation de fer [20]. Les cytokines
inflammatoires agissent également directement au niveau
des progéniteurs érythroïdes, inhibent leur différenciation
et leur prolifération [21] en provoquant leur apoptose ou
en produisant des radicaux libres qui altèrent les capaci-
tés de liaison de l’érythropoïétine (EPO) à ses récepteurs.
Le TNF␣augmente l’apoptose des cellules érythroïdes et
l’interféron gamma semble être l’inhibiteur le plus puissant
de l’érythropoïèse.
Ces phénomènes conduisent à une diminution du fer dis-
ponible, avec hyposidérémie, alors que les stocks martiaux
sont intacts, voire augmentés. Cette carence martiale rela-
tive (ou fonctionnelle) associée à une séquestration martiale
peut être visualisée par la coloration de Perls au niveau des
cellules réticuloendothéliales de la moelle osseuse : on y
observe une absence de fer érythroblastique (sidéroblastes)
et la présence de fer dans le système réticulo-endothélial
(macrophages). Le dosage des TfRs est corrélé à ces anoma-
lies médullaires, en présentant un taux normal ou diminué.
L’anémie inflammatoire au cours de la pathologie can-
céreuse peut par ailleurs être associée aux autres causes
d’anémie comme les carences en fer par saignement
occulte, l’hémolyse, les métastases médullaires ou la myé-
losupression induite par les chimiothérapies. Le diagnostic
différentiel est important pour une prise en charge adap-
tée. Il repose sur le bilan biologique comprenant un
hémogramme, le dosage du fer plasmatique, le CST, la
ferritinémie et le dosage de marqueurs de l’inflammation
(tableau 1).
Les anomalies du métabolisme du fer
dans le cancer du sein
Modèles précliniques
Surcharge en fer des cellules tumorales mammaires
Le pouvoir carcinogène du fer a été démontré dans de nom-
breux modèles animaux et cellulaires. Le rôle du fer est
crucial pour le fonctionnement de ribonucléotide-réductase
(RR), l’enzyme qui génère les nucléotides pour la syn-
thèse et la réparation de l’ADN. Les cellules tumorales ont
des besoins en fer augmentés car proliférant rapidement.
L’exposition des cellules en division aux chélateurs de fer
peut conduire à l’arrêt du cycle cellulaire. Des études pré-
cliniques portant sur la séquestration du fer par l’utilisation
d’agents chélateurs du fer ont d’ores et déjà été rapportées
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Ann Biol Clin, vol. 70, n◦4, juillet-août 2012 391
Fer et cancer du sein
Tableau 1. Diagnostic biologique des anémies inflammatoires au
cours des cancers (cas d’une anémie inflammatoire pure sans
carence martiale associée).
Hémogramme
Anémie
généralement modérée, de 90 et 110 g/L,
normochrome, normocytaire, arégénérative
pouvant évoluer vers une forme microcytaire modérée
(70 >VGM >80 fL)
Hyperleucocytose avec polynucléose neutrophile
Thrombocytose
Bilan martial
Fer sérique bas
CST abaissé ou normal
CTF normale ou diminuée
Ferritinémie élevée
Ferritine érythrocytaire normale
Récepteur soluble de la transferrine normal
Biologie du syndrome inflammatoire
Vitesse de sédimentation augmentée
Fibrinogène augmenté
Hyper-gamma et hyper-alpha-2-globulinémie
Haptoglobine et CRP élevées
[22]. Le chélateur généralement utilisé en pratique clinique
dans le traitement des surcharges en fer, la déféroxamine
(DFO) [23], a démontré dans les années 1980 des proprié-
tés antitumorales, tant in vitro [22, 24] que in vivo [22] ainsi
que dans le cadre d’essais cliniques du neuroblastome [25].
La DFO agit par déplétion du pool labile de fer, nécessaire
à l’activité enzymatique de la RR. Toutefois, l’efficacité de
la DFO est limitée en raison de sa faible capacité à traver-
ser les membranes cellulaires et à lier le fer du pool labile.
Elle ne s’est pas révélée efficace dans des modèles murins
de leucémie aiguë myéloblastique et de carcinome du col
utérin [26] et en clinique dans la prise en charge du can-
cer de la prostate [27]. Cependant, récemment une équipe
japonaise a rapporté des résultats encourageants en utilisant
de la DFO en perfusion intra-artérielle hépatique chez 10
patients atteints d’un carcinome hépatocellulaire. Le taux
de réponse partielle était de 20 %, avec 30 % de stabilisation
tumorale [28].
Les limites de la DFO ont encouragé le développement
d’autres chélateurs plus efficaces. Les investigations se sont
notamment portées sur un groupe de ligands de la classe
des PIH (pyridoxal isonicotinoyl hydrazone). Ces agents
ont montré une plus grande capacité de chélation que la
DFO. En effet, les PIHont montré une efficacité cytotoxique
comparable à celle de la bléomycine et du cisplatine dans
des modèles précliniques [29]. Cependant nous ne dispo-
sons pas actuellement d’études cliniques venant confirmer
ces données chez l’homme. Les chélateurs de fer ont été
utilisés in vitro sur les lignées cellulaires de cancer du
sein et ont montré une certaine activité cytostatique [30].
En plus de la déplétion en fer, certains chélateurs de fer
peuvent avoir une activité antitumorale via une inhibition
de l’activité de la topo-isomérase II ␣[31].
De fac¸on plus générale, les cellules cancéreuses présentent
un déséquilibre de la balance en fer en faveur d’une éléva-
tion de la concentration intracellulaire de Fe3+ [32, 33]. Il
a été montré qu’une une diète appauvrie en fer provoque
chez le rat une diminution de la croissance des cancers de
la glande mammaire [34].
L’étude de Pinnix et al. [1] récemment publiée met en
lumière le rôle important que jouent la ferroportine et
l’hepcidine dans les cancers du sein. Les auteurs ont tout
d’abord montré que la ferroportine et l’hepcidine sont expri-
mées par les cellules cancéreuses mammaires en culture
et que l’hepcidine régule négativement par un mécanisme
post-transcriptionnel l’expression de la ferroportine dont
les taux dans les cellules tumorales sont diminués par rap-
port aux cellules normales. La concentration en hepcidine
est par ailleurs plus élevée dans les cellules tumorales que
dans les cellules normales. L’augmentation du ratio hepci-
dine/ferroportine conduit à une augmentation de la ferritine
et du fer labile dans les cellules cancéreuses.
Plusieurs études ont montré une augmentation de la ferri-
tine tissulaire dans les tumeurs solides dont le cancer du sein
[35]. Les altérations de l’expression de la ferritine ont été
retrouvées dans les lignées cellulaires du cancer du sein,
mais aussi sur les tissus d’hyperplasie canalaire atypique
et de carcinome canalaire in situ, ce qui laisse penser que
la régulation positive du gène humain de la ferritine (FTH)
intervient dans les étapes précoces de la carcinogenèse [36].
La surexpression du FTH a été retrouvée sur les lignées cel-
lulaires du cancer du sein de phénotype mésenchymateux
agressif [37]. Le micro-RNA 200b régule négativement le
FTH et conduit à une meilleure réponse in vitro à la doxo-
rubicine [37].
Le fer, un oxydant de l’ADN
Le fer est à la fois un élément indispensable de la vie cel-
lulaire, notamment au niveau de la respiration, mais aussi
un élément toxique qui peut induire un stress oxydatif et
des altérations de l’ADN. La mitochondrie produit à partir
d’une partie de l’oxygène des espèces réactives oxygénées
(ROS) comme l’anion superoxyde (O2˙-) et le peroxyde
d’oxygène (H2O2). Ces derniers sont transformés par la
superoxyde dismutase (SOD), la catalase (CAT) et la glu-
thatione peroxydase (GPx) en eau. En présence d’anion
superoxyde et de peroxyde d’hydrogène, le Fe3+ est réduit
en Fe2+ qui catalyse (réactions de Fenton et Haber-Weiss)
la production de radicaux hydroxyl très actifs capables de
former des adduits au niveau de l’ADN et d’initier la car-
cinogenèse [38].
Le processus de la peroxydation des lipides produit du
MDA (malondialdéhyde) qui interagit avec les bases gua-
nine, adénine et cytosine de l’ADN. Les produits de
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%