Apport de la génétique dans la prise en charge des surdités

L’
identification des gènes responsables de surdité a été
menée tambour battant ces dernières années (le pre-
mier gène a été localisé en 1993), et comme plusieurs
dizaines de gènes sont vraisemblablement impliquées, elle n’est
pas finie. Mais devant la multiplication des connaissances fon-
damentales, une question s’impose : quel est l’intérêt de ces
découvertes pour le praticien ?
En fait, les recherches génétiques ont remis en valeur le diagnostic
clinique. Elles ont permis de définir des points essentiels sur les-
quels l’interrogatoire doit porter pour identifier une surdité géné-
tique, même en l’absence d’antécédent familial de surdité. Un
nombre restreint, mais essentiel, d’examens complémentaires
doit être demandé. Enfin, une caractérisation clinique soigneuse
est nécessaire avant d’envisager une analyse génétique, qui est
réservée à certains cas.
GÉNÉTIQUE ET DIAGNOSTIC DES SURDITÉS
Génétique clinique
Quand proposer une enquête génétique ?
Une cause génétique mérite d’être recherchée dans de nom-
breux cas.
En effet, les causes génétiques sont responsables de plus de 50 %
des surdités de perception de l’enfant. Une atteinte génétique peut
également provoquer une malformation de l’oreille externe ou
des osselets. Chez l’adulte, le facteur génétique intervient dans
l’otospongiose, la presbyacousie, l’ototoxicité des aminosides,
et dans certains cas de maladie de Ménière (1-7).
Recherche d’antécédents familiaux
Rechercher l’existence de cas familiaux de surdité est néces-
saire devant toute surdité, mais non suffisant. En effet, une cause
génétique peut exister en l’absence d’antécédents familiaux de
surdité (tableau I).
Il faut également poser la question des pathologies formant un
syndrome génétique connu avec la surdité (diabète, mèche
blanche, rétinite pigmentaire, etc.) (tableau II), car les signes
d’un syndrome peuvent être dispersés chez des personnes diffé-
rentes d’une même famille. De plus, une consanguinité, une
même origine géographique des familles des deux parents
(consanguinité éloignée), ou un âge parental avancé (en faveur
d’une mutation de novo), sont des éléments d’orientation.
Enfin, il est utile de pratiquer un audiogramme chez les parents
et les frères et sœurs du sujet malentendant, car une perte audi-
tive discrète peut être ignorée.
S’il existe des antécédents familiaux de surdité, deux étapes sont
nécessaires avant d’affirmer le caractère génétique de la surdité :
tout d’abord vérifier l’absence de cause acquise de surdité (trau-
matisme sonore, méningite, ototoxiques, etc.) chez les diverses
personnes atteintes ; ensuite, déterminer le mode de transmission.
La répartition des différents modes de transmission des surdités
est représentée sur la figure 1 (8).
Recherche d’un syndrome associé à la surdité
C’est un élément majeur du diagnostic, car 30 % des surdités
génétiques s’intègrent dans un syndrome (1-3). Cette recherche
repose avant tout sur un interrogatoire détaillé concernant les
DOSSIER
Apport de la génétique dans la prise en charge
des surdités
●
G. Lina-Granade*, E. Truy*, N. Alloisio**, L. Morlé**, F. Disant***, L. Collet*, P. Edery***, H. Plauchu*****
17
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - no261 - mars 2001
* Département d’ORL, de chirurgie cervico-faciale et d’audiophonologie et
Laboratoire neurosciences et systèmes sensoriels, UPRESA CNRS 5020,
hôpital Édouard-Herriot, Lyon.
** Centre de génétique moléculaire et cellulaire, université Claude-Bernard-
Lyon-I, Villeurbanne.
*** Département d’ORL, de chirurgie cervico-faciale et d’audiophonologie,
hôpital Édouard-Herriot, Lyon.
**** Centre de génétique moléculaire et cellulaire, université Claude-Bernard-
Lyon-I, Villeurbanne et service génétique, Hôtel-Dieu, Lyon.
***** Service de génétique, Hôtel-Dieu, Lyon.
Tableau I. Facteurs expliquant l’absence fréquente d’antécédent
familial de surdité dans les surdités génétiques.
Facteur explicatif
Fréquence
des formes récessives
Expression variable
des syndromes
Pénétrance incomplète
Mutation
de l’ADN mitochondrial
Surdités évolutives
Explication
Les parents, frères et sœurs sont porteurs
du gène, mais normo-entendants
La surdité n’est pas présente
chez tous les sujets atteints, qui présentent
d’autres signes du syndrome
Les sujets porteurs du gène ne manifestent
pas toujours la maladie
(par exemple, l’otospongiose)
Ces surdités génétiques apparaissent
parfois après un événement extérieur
(prise d’aminosides)
Les personnes devenant malentendantes
ne le signalent pas toujours à leur entourage

atteintes les plus fréquemment associées à une surdité génétique
(tableau II). En dehors de cette brève liste, de nombreux syn-
dromes plus rares comportent une surdité, mais elle n’est pas au
premier plan.
Outre l’interrogatoire, on examinera donc le visage, le cou, les
oreilles. On demandera, chez l’enfant, un examen ophtalmolo-
gique avec fond d’œil, une recherche de protéinurie et hématu-
rie par bandelette urinaire, et un électrocardiogramme par un spé-
cialiste de cardiologie pédiatrique.
Chez l’adulte, le bilan de toute surdité de perception comprend
une glycémie à jeun, voire une épreuve d’hyperglycémie provo-
quée en cas d’antécédents maternels de diabète.
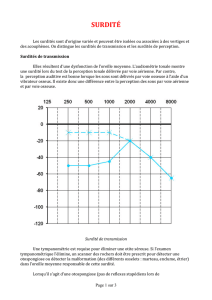
Caractérisation de la surdité et radiologie
Hormis l’otospongiose, la plupart des surdités génétiques sont
des surdités de perception prédominant sur les aigus. Les aspects
audiométriques typiques (surdité de perception prédominant sur
les graves ou les médiums) sont rares.
Une atteinte vestibulaire doit être systématiquement recherchée
par l’interrogatoire, car elle s’associe à certaines formes de sur-
dité génétique.
Quel que soit l’âge, le scanner des rochers permettra de dé-
tecter les malformations de l’oreille interne. En particulier, la
DOSSIER
18
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - no261 - mars 2001
Tableau II. Principaux syndromes génétiques comprenant une surdité.
Diabète
Pigmentation
(peau,
phanères,
iris)
Oreille
externe
et face
Rétine
Reins
Thyroïde
Conduction
cardiaque
Nom
du syndrome
Surdité-diabète
mitochondrial
Syndrome
de Waardenburg
Syndrome
branchio-oto-rénal
Syndrome
de Franceschetti
ou Treacher-Collins
(dysostose
mandibulo-faciale)
Syndrome d’Usher
Syndrome d’Alport
Syndrome de Pendred
Syndrome de Jervell-
Lange-Nielsen
Transmission
familiale
Maternelle (mutation
mitochondriale)
Autosomique dominant,
àpénétrance incomplète
et expressivité
très variable
Autosomique dominant
àexpressivité variable,
avec signes parfois
discrets
Autosomique dominant
àexpressivité variable
et pénétrance incomplète
Autosomique récessif
Le plus souvent dominant lié
àl’X (grave chez l’homme,
bénin chez la femme)
Autosomique récessif
Autosomique récessif
Caractéristiques
de la surdité
Surdité de perception
progressive
Surdité de perception précoce,
de gravité variable,
uni- ou bilatérale
Surdité de perception,
de transmission
ou mixte
Surdité de transmission
Surdité congénitale, moyenne
àprofonde, stable
Surdité de perception
progressive,
débutant à l’adolescence
Surdité de perception
congénitale, bilatérale
et sévère
Surdité de perception
congénitale, sévère ou profonde
Autres signes
Diabète non insulino-dépendant, débutant avant 40 ans,
devenant insulino-dépendant, de transmission maternelle
• Albinisme partiel (yeux vairons, mèche de cheveux
blancs, blanchissement précoce des cheveux,
taches cutanées non pigmentées)
• Parfois écartement excessif des yeux
• Pavillon de l’oreille : anomalie de l’anthélix, microtie
• Orifice préauriculaire ou fistule branchiale
• Appareil urinaire : agénésie rénale,
duplication pyélique, défaut de rotation, etc.
• Oreilles externes : microtie, atrésie du conduit auditif
externe
• Face “longue”: micrognathie, hypoplasie
des malaires, fentes palpébrales obliques
avec colobome (fissure) de la paupière inférieure
Rétinite pigmentaire (baisse visuelle ou rétrécissement
du champ visuel, dans l’enfance ou l’adolescence)
Glomérulonéphrite hématurique, débutant
dans l’enfance, évoluant vers l’insuffisance rénale
àl’adolescence chez l’homme
• Goitre euthyroïdien à l’adolescence
• Dilatation de l’aqueduc du vestibule,
hypoplasie cochléaire
• Syncopes dans l’enfance
• Intervalle Q-T long à l’ECG
Figure 1. Modes de transmission des surdités génétiques.
Non syndromiques
liées à l’X
1 %
Non syndromiques
dominantes
Non syndromiques
autosomiques récessives
65 %
10 %
Syndromiques
20 à 30 %
.../...

DOSSIER
20
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - no261 - mars 2001
Tableau III. Principaux gènes responsables de surdité non syndromique et tableaux cliniques correspondant (12, 13). Les gènes de surdité DFN
sont liés à l’X, les gènes DFNA sont autosomiques dominants, les gènes DFNB sont autosomiques récessifs.
Transporteurs
ou canaux
(homéostasie
endolymphe)
Cytosquelette
Facteurs
de
transcription
Matrice
extracellulaire
Métabolisme
oxydatif
Nom
de la
protéine
Connexine 26
(GJB2)
Connexine 31
(GJB3)
Connexine 30
(GJB6)
Pendrine
KCNQ4
Myosine VIIA
Myosine XV
Diaphanous
Otoferline
POU3F4
POU4F3
Tectorine
COCH
Fraction
ribosome 12S
ARNt
de la sérine
Chromosome
13q12
1p32
13q12
7q31
1p34
11q13
17p11
5q31
2p23
Xq21
5q31
11q23
14q12
Mitochondrial
np1555
Mitochondrial
np7445
Nom
du
gène
DFNB1
DFNA3
DFNA20
DFNA3
PDS
DFNB4
DFNA2
USH IB
DFNB2
DFNA11
DFNB3
DFNA1
DFNB9
DFN3
DFNA15
DFNA12
DFNB21
DFNA9
Caractéristiques
de la surdité
de perception non syndromique
• Récessive : congénitale modérée
àprofonde, plate ou sur aigus,
sans anomalie radiographique
• Dominante : modérée à sévère,
prélinguale, progressive,
sans atteinte vestibulaire
Progressive, débutant entre 20 et 40 ans,
légère-moyenne, sur aigus,
avec acouphène, de pénétrance incomplète
Progressive, légère à profonde,
sur médiums et aigus
Congénitale, profonde,
avec dilatation de l’aqueduc
du vestibule
Progressive, postlinguale, sur aigus
• Récessive : profonde, +/– aréflexie
vestibulaire, congénitale
ou début avant 16 ans
• Dominante : modérée, progressive,
début entre 4 et10 ans,
sans atteinte vestibulaire
Profonde, congénitale,
sans trouble vestibulaire
Progressive, début vers10 ans,
profonde à 30 ans
Sévère à profonde, prélinguale
Progressive, mixte, avec fixation de l’étrier
et abouchement CAI-cochlée
Progressive, début 18-30 ans,
modérée à sévère,
avec atteinte vestibulaire
Prélinguale, stable ou progressive,
légère à sévère
Progressive, début 35-55 ans,
avec instabilité +/–épisodes méniériformes
Prélinguale ou progressive
(enfant ou adulte), légère à profonde,
parfois après aminosides,
sans atteinte vestibulaire
Début entre 3 et 18 ans, progressive,
légère à moyenne
Nombre de familles
publiées, origine
géographique
Forme récessive
la plus fréquente
(Europe, Afrique,
États-Unis, Pakistan,
etc.)
2 (Chine)
1 (Italie)
2 (Proche-Orient,
Inde)
6 (Europe,
États-Unis)
Tunisie, Chine,
Japon
3 (Bali, Inde)
1 (Costa Rica)
4 (Liban)
Forme liée à l’X
la plus fréquente
1 (Sépharades)
3 (France, Belgique,
Autriche)
États-Unis
Belgique, Hollande
20 % des surdités
familiales ?
Europe, Afrique, Asie
3 (Écosse, Japon,
Nouvelle-Zélande)
Expression
et rôle
de la protéine
Spécifique des cellules
cochléaires
non sensorielles :
canaux intercellulaires
pour le recyclage du K
Idem
Canaux intercellulaires,
dans cochlée, cerveau,
trachée, thyroïde
Transporteur
transmembranaire
des iodures (thyroïde)
et chlorures (cochlée)
Cerveau, cellules ciliées
externes
Cellules épithéliales ciliées
(rétine, CCI) : transport
intracellulaire
Cochlée (cils), cerveau,
gonades, rein
Ubiquitaire : polarité
cellulaire, polymérisation
de l’actine (cils)
Spécifique des CCI et des
cellules vestibulaires I :
vésicules, membrane
Cerveau, tube neural,
vésicule otique embryon
Spécifique des cellules
ciliées cochléaires
(fœtus et adulte) :
différenciation, survie
Membrane tectoriale
(spécifique de la cochlée)
Conjonctif et nerf
cochléo-vestibulaires
Ubiquitaire,
mitochondries
Ubiquitaire,
mitochondries
.../...

21
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - no261 - mars 2001
découverte d’une dilatation de l’aqueduc du vestibule (DAV) ou
d’une hypoplasie cochléaire (Mondini) oriente vers une cause
génétique (9).
Le scanner est indispensable devant toute surdité mixte à réflexe
stapédien aboli, afin de distinguer otospongiose et “oreille gey-
ser”. Cette dernière est caractérisée par une dilatation ampullaire
du conduit auditif interne, et une fistule entre conduit auditif
interne et tour basal de la cochlée (10).
En cas de malformation de l’oreille externe associée à un kyste
ou à une fistule branchiale, il est prudent de réaliser une écho-
graphie rénale à la recherche d’une malformation liée au syn-
drome branchio-oto-rénal.
GÉNÉTIQUE MOLÉCULAIRE
Le caryotype est sans intérêt en l’absence d’un syndrome mal-
formatif et d’un retard mental, car il ne permet d’identifier que
les anomalies chromosomiques (trisomie 21, syndrome de Tur-
ner, translocations, etc.), qui s’accompagnent d’une surdité dans
certains cas.
L’analyse du gène de la connexine 26 peut être proposée quand
la surdité est isolée (sans syndrome), congénitale, quel que soit
son degré, et si la généalogie évoque une surdité récessive (parents
normo-entendants). En effet, les mutations de ce gène semblent
responsables de près de 50 % des surdités récessives isolées (11).
Il est donc intéressant d’analyser ce gène, même quand il n’y a
pas d’antécédent familial. En revanche, si l’analyse de la
connexine 26 ne met pas en évidence de mutation, comme plus
de 50 gènes sont impliqués dans les surdités, une cause génétique
ne peut être exclue (12, 13).
En cas d’association surdité et diabète de transmission mater-
nelle, la recherche d’une mutation de l’ADN mitochondrial peut
être réalisée dans certains laboratoires hospitaliers de biochimie
ou biologie moléculaire.
Actuellement, il s’agit des seuls gènes analysables en routine,
bien que d’autres gènes soient identifiés (tableau III). Les gènes
impliqués dans les syndromes présentés dans le tableau II sont
pour la plupart identifiés ; l’analyse n’est pas réalisée couram-
ment, mais comme ces données évoluent rapidement, nous
conseillons de consulter le serveur Orphanet (14).
Enfin, les familles dans lesquelles le diagnostic de surdité géné-
tique est bien établi (nombreux sujets atteints, élimination d’une
cause acquise, mode de transmission caractérisé) peuvent par-
ticiper à l’identification de gènes de surdité encore inconnus,
et aider à évaluer la répartition des divers gènes dans la popu-
lation française. Les familles atteintes doivent donc être réfé-
rées aux centres pratiquant ces recherches, indiqués sur le site
Orphanet (14).
GÉNÉTIQUE DANS LE SUIVI DES SURDITÉS
Conseil génétique et détection des porteurs sains
Le diagnostic de surdité génétique permet de proposer aux
familles un conseil génétique, avec estimation du risque pour un
autre membre de la famille d’être sourd. Il ne s’agit que d’une
estimation, car le Comité national d’éthique a recommandé de ne
pas pratiquer de diagnostic anténatal pour les surdités. Les argu-
ments contre le diagnostic anténatal sont qu’une même mutation
peut provoquer des degrés de surdité très divers (11), et qu’un
enfant sourd peut bénéficier d’une prise en charge précoce per-
mettant de compenser son handicap, en particulier par implanta-
tion cochléaire.
Le risque de transmission de la surdité aux enfants varie de 100 %
pour une femme atteinte de surdité mitochondriale, à 25 % pour
deux parents porteurs d’un gène récessif.
Pour un patient atteint d’une surdité récessive, le risque d’avoir un
enfant sourd dépend du conjoint, et l’analyse de la connexine 26
est alors conseillée. En effet, la fréquence des gènes de surdité dans
la population est très élevée : une personne sur huit (15 % de la
population) serait porteuse d’un gène de surdité récessive, et une
personne sur trente (2,5 à 4 %) d’une mutation récessive de la
connexine 26 (15).
Enfin, lorsque l’on détecte une mutation de la connexine 26, le
généticien peut proposer l’analyse génétique aux frères et sœurs
des sujets atteints et des porteurs sains, pour déterminer s’ils sont
également porteurs de la mutation.
Pronostic évolutif et règles hygiéno-diététiques
Le diagnostic de surdité génétique permet dans certains cas
typiques d’apprécier le risque évolutif, en fonction des données
connues concernant chaque forme de surdité génétique
(tableau III). Toutefois, l’évolutivité peut varier selon les
patients, et la surveillance audiométrique annuelle du patient
concerné est le meilleur moyen d’établir le pronostic pour lui.
Comme devant toute surdité de perception potentiellement évo-
lutive, on prendra en compte tout facteur pouvant accélérer la
perte auditive : diabète, hypertension, troubles lipidiques, trau-
matismes sonores, barotraumatismes, médicaments ototoxiques,
etc. En cas de dilatation de l’aqueduc du vestibule, les activités
présentant un risque de traumatisme crânien et les efforts vio-
lents seront déconseillés.
Par ailleurs, l’appareillage auditif précoce permettra de limiter
la dégénérescence nerveuse et la détérioration de l’intelligibilité
de la parole. Enfin, il faut conseiller aux patients de consulter en
urgence si une perte auditive brusque se produit, afin de mettre
en route rapidement un traitement corticoïde et vasodilatateur.
Compréhension de la physiopathologie
des surdités génétiques
L’identification des gènes responsables de surdité a permis de
découvrir des protéines jouant un rôle important dans la struc-
ture et la fonction de la cochlée (tableau III). Une surdité géné-
tique peut ainsi être due aussi bien à une dysfonction des cellules
sensorielles qu’à un déficit du recyclage du potassium dans
l’endolymphe (en cas d’atteinte de la connexine 26) ou à une ano-
malie de la membrane tectoriale.
L’identification des gènes de surdités génétiques a apporté de
nouvelles preuves que la transmission familiale et les symptômes
ne dépendent pas seulement du gène impliqué, mais de la muta-
tion que porte ce gène : en effet, un même gène peut être res-
ponsable, selon les mutations, d’une surdité récessive ou domi-

DOSSIER
22
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - no261 - mars 2001
nante (gène de la connexine 26), ou d’une surdité isolée ou d’un
syndrome (gène de la myosine VIIA).
Perspectives thérapeutiques
La thérapie génique dans les surdités n’a été envisagée jusqu’ici
que chez l’animal. Un premier axe de recherche consiste à trans-
férer un gène “correcteur” chez des individus présentant une
mutation de ce gène responsable de surdité. Malheureusement,
les gènes impliqués dans les surdités génétiques agissent très pré-
cocément au cours du développement de la cochlée. Le transfert
du gène de la surdité après la naissance ne permettrait donc pas
de corriger le déficit auditif. Les expériences déjà publiées por-
tent ainsi sur l’effet préventif du transfert de gène chez l’embryon,
ou dans les cellules germinales des parents (16).
Une deuxième voie porte sur le transfert, dans l’oreille interne
de l’animal adulte, de gènes de facteurs de croissance neuronaux
(BDNF, GDNF) afin d’éviter la destruction des cellules ciliées
et des neurones provoquée par les aminosides (17). L’intérêt de
ces gènes dans les surdités génétiques reste à déterminer.
CONCLUSION
L’amélioration du diagnostic clinique des surdités génétiques et
l’identification de gènes responsables permettent de répondre
dans certains cas aux questions posées par les patients : “Pour-
quoi suis-je malentendant ? Comment cette surdité va-t-elle évo-
luer ? Quel est le risque de surdité pour mes enfants, mes frères
et sœurs”, etc. ?
En revanche, il n’existe pas encore de traitement préventif, voire
curatif pour ces affections. C’est pourquoi les facteurs pouvant
aggraver la surdité (facteurs métaboliques et traumatiques en par-
ticulier) et l’appareillage auditif précoce doivent toujours être
envisagés pour limiter la dégradation auditive.
Enfin, les surdités génétiques n’ont pas livré tous leurs secrets,
et la poursuite des recherches est nécessaire pour identifier les
gènes les plus fréquents (à côté du gène de la connexine 26) et
l’effet de l’association de plusieurs mutations.
■
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Martini A, Read A, Stephens D. Genetics and hearing impairment. Whurr
Publishers : Londres, 1996.
2. Lina-Granade G, Plauchu H, Morgon A. Les surdités génétiques. Mono-
graphies du CCA Wagram : Paris, 1995.
3. Gorlin RJ, Toriello HV, Cohen MM. Hereditary hearing loss and its syn-
dromes. Oxford University Press : Oxford, 1995.
4. Higashi K et al. Familial ossicular malformations : case report and review
of the literature. Am J Med Genet 1987 ; 28 : 655-9.
5. Lina-Granade G, Collet L, Morgon A. Presbyacousie: génétique, examens
audiologiques et prise en charge médicale. Lettre d’ORL 1996 ; 206 : 4-6.
6. Fischel-Ghodsian N. Mitochondrial deafness mutations reviewed. Hum
Mutation 1999 ; 13 : 261-70.
7. Martini A. Hereditary Meniere’s disease : report of two families. Am J
Otolaryngol 1982 ; 3 : 163-7.
8. Willems JW. Genetic causes of hearing loss. N Engl J Med 2000 ; 342 :
1101-9.
9. Usami S, Abe S, Weston MD et al. Non syndromic hearing loss associated
with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet
1999 ; 104 : 188-92.
10. Phelps P, Reardon W, Pembrey ME, Bellman S, Luxon L. X-linked deaf-
ness, stapes “gusher” and a distinctive defect of the inner ear. Neuroradiol
1991 ; 33 : 326-30.
11. Denoyelle F, Marlin S, Weil D et al. Clinical features of the prevalent
form of childhood deafness DFNB1, due to a connexin-26 gene defect : impli-
cations for genetic counselling. Lancet 1999 ; 353 : 1298-303.
12. Van Camp G, Smith RJH. Hereditary hearing loss homepage. http:
//dnalab-www.uia.ac.be/dnalab/hhh.html.
13. Lina-Granade G, Morlé L, Alloisio N et al. Les surdités génétiques, pre-
mière cause de surdité de perception de l’enfant. Arch Pediatr 2001 (sous
presse).
14. Annuaire des maladies rares Orphanet. Éditions INSERM : Paris, 1998,
ou http://orphanet.infobiogen.fr
15. Estivill X, Fortina P, Surrey S et al. Connexin-26 mutations in sporadic
and inherited sensorineural deafness. Lancet 1998 ; 351 : 394-8.
16. Probst KJ, Fridell RA, Raphael Y et al. Correction of deafness in shaker-
2 mice by an unconventional myosin in a BAC transgene. Science 1998 ; 280 :
1444-7.
17. Yagi M, Magal E, Sheng Z et al. Hair cell protection from aminoglyco-
side ototoxicity by adenovirus-mediated overexpression of GDNF. Hum Gene
Therapy 1999 ; 10 : 813-23.
Voir aussi rubrique Internet p. 12-13
Brèves Internet
Un nouvel annuaire électronique
Sur le site www.1bis.com, on peut non seulement rechercher les
coordonnées d’une entreprise ou d’un ami, mais également
retrouver un nom à partir du numéro de téléphone. De plus, il
existe un service très pratique : en tapant le nom d’une rue et
d’une ville, le plan s’affiche. Vous pouvez l’enregistrer, l’expé-
dier par mail, obtenir des précisions sur les accès par les trans-
ports en commun. Ce site concerne toutes les grandes villes de
France et sera bientôt pourvu d’un calculateur d’itinéraires.
R. Marianowski
1
/
5
100%