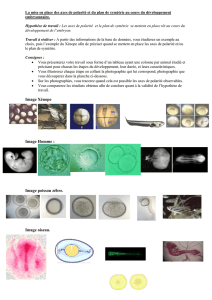
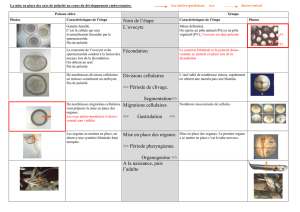
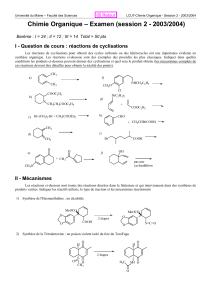
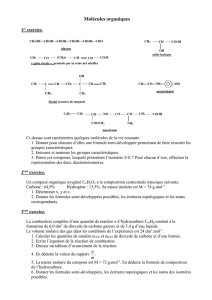
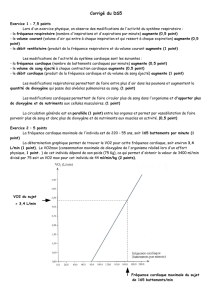
Analyse rétrosynthétique - Cours de chimie générale

Analyse
rétrosynthétique

La synthèse vers l'arrière
Termes utilisés:
Molécule cible: molécule à synthétiser
Déconnexion: coupure imaginaire d'une liaison, correspondant à
l'inverse d'une réaction réelle
Synthon: fragment schématique résultant d'une déconnexion,
remplacé par un réactif dans la synthèse
Equivalent synthétique (ES): réactif ayant les propriétés
caractéristiques d'un synthon
OH
+
O

1.1. Choix d'une déconnexion
Concepts de base
Les déconnexions doivent correspondre à des réactions connues et
fiables.
Les composés constitués de deux parties reliées par un hétéroatome
sont déconnectés à côté de l'hétéroatome.
Choisir les voies qui évitent les problèmes de chimiosélectivité, cela
signifie en général qu'il faut déconnecter d'abord les groupements
réactifs.
Les déconnexions de deux groupes sont meilleures que celles d'un
seul.

Exemple 1: réalisez l'analyse rétrosynthétique de la cible suivante:
Cl
O
Cl
COOH

Exemple 2: réalisez l'analyse rétrosynthétique de la cible suivante:
N
H
COOEt
R
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
1
/
35
100%