Les canalopathies cardiaques

doi: 10.1684/epi.2010.0326
Les canalopathies cardiaques
Isabelle Denjoy
1,2,3
, Jean-Marc Lupoglazoff
2,3
, Fabrice Extramiana
1
,
Antoine Leenhardt
1
1
AP-HP, Hôpital Lariboisière, service de cardiologie, Paris, F-75010, France
2
AP-HP, Hôpital Robert Debré, Service de cardiopédiatrie, Paris, F-75019, France
3
Inserm, UMR_S 956, Faculté de médecine Pitié-Salpétrière, Paris, F-75013, France
Résumé. Les arythmies ventriculaires cardiaques malignes primitives sont rares mais exposent les sujets qui
en sont atteints à des syncopes ou à la mort subite, souvent à l’occasion d’une stimulation adrénergique. Elles sont
dues à des anomalies de fonctionnement d’origine génétique de certains canaux ioniques cardiaques (canalo-
pathie). Parmi ces arythmies ventriculaires malignes, dues à des anomalies de fonctionnement primitif des canaux
ioniques cardiaques, on distingue le syndrome du QT long (dysfonction de canaux potassiques KCNQ1, KCNH2,
ou sodique SCN5A), le syndrome de Brugada (dysfonction du canal sodique SCN5A), le syndrome du QT court, et
les tachycardies ventriculaires catécholergiques (dysfonction du récepteur à la Ryanodine). L’expression clinique est
la syncope, souvent accompagnée de convulsions ou l’arrêt cardiaque par troubles du rythme ventriculaire graves
(tachycardie ventriculaire polymorphe, torsades de pointes, fibrillation ventriculaire). Le dépistage cardiologique
de l’un de ces syndromes repose sur l’analyse de l’ECG, les antécédents personnels de chaque membre de la famille,
éventuellement les tests pharmacologiques de sensibilisation. Pour certains de ces syndromes, le ou les gènes en
cause sont connus et rendent possible, au sein d’équipes multidisciplinaires spécialisées, la prise en charge des
apparentés présymptomatiques ou symptomatiques dont le diagnostic n’aurait pas été établi. Toute mort subite
rythmique récupérée expose le patient à un risque élevé de récidive et justifie l’implantation d’un défibrillateur
automatique.
Mots clés :mort subite,syncope,génétique,arythmies ventriculaires,canalopathies
Abstract. Cardiac channelopathies
Primary ventricular arrhythmias are rare but affected patients carry a high risk of syncope or sudden death by torsade
de pointes or polymorphic ventricular tachycardia... Primary ventricular arrhythmias are due to cardiac channelopa-
thies genetically determined. The main channelopathies include: the long QT syndrome (potassium KCNQ1 and
KCNH2 channelopathies and cardiac sodium SCN5A channelopathy), Brugada syndrome (cardiac sodium channel
SCN5A channelopathy), short QT syndrome and Polymorphic Ventricular Tachycardia (Ryanodine receptor channe-
lopathy). Clinical manifestations which include syncope with eventually seizure and cardiac arrest are mainly
adrenergically-triggered. The diagnosis of such syndromes relies upon specific ECG anomalies and stress test if
necessary, personal history of family members, eventually echocardiography and drug challenge. For some of these
diseases, morbid genes have been identified thus rendering possible the management of pre symptomatic or
undiagnosed family members within specialized multidisciplinary teams. In cases of sudden arrhythmic death in
children, the parents and siblings must be examined Rescued sudden death exposes to a high risk of recurrence. In
such patients, the automatic implantable defibrillator has dramatically improved survival.
Key words:sudden death,syncope,genetics-,ventricular arrhythmias,channeloptahies
Épilepsie et coeur
Épilepsie et coeur
Épilepsies 2010 ; 22 (3) : 230-4
Tirés à part :
I. Denjoy
Épilepsies, vol. 22, n° 3, juillet-août-septembre 2010 230
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Les arythmies ventriculaires malignes primitives sont rares
mais exposent les sujets qui en sont atteints à des syncopes
ou à la mort subite (MS), souvent à l’occasion d’une stimulation
adrénergique. Elles sont dues à des anomalies de fonctionne-
ment d’origine génétique de certains canaux ioniques cardia-
ques (canalopathie). Ces anomalies rythmiques primitives
incluent les torsades de pointes dégénérant en fibrillation
ventriculaire pour le syndrome du QT long congénital, les
tachycardies ventriculaires catécholergiques (TVC), les fibrilla-
tions ventriculaires compliquant le syndrome de Brugada ou le
syndrome du QT court. L’origine génétique de ces syndromes
en fait des maladies héréditaires et familiales. La confirmation
du diagnostic est possible en cas d’identification de l’anomalie
génétique chez le sujet atteint et chez les membres de sa
famille. Le traitement par bêtabloquants et/ou le défibrillateur
automatique implantable (DAI), ainsi qu’une restriction
majeure de la pratique sportive contribuent à l’amélioration
du pronostic vital.
Syndrome du QT long congénital
Diagnostic
Le syndrome du QT long congénital (QTL), cliniquement
et génétiquement hétérogène, se caractérise par un allonge-
ment de l’intervalle QT sur l’ECG de surface (QTc > 440 ms),
associé à un risque élevé de survenue de troubles du rythme
ventriculaire graves (torsades de pointes, fibrillation ventricu-
laire) pouvant entraîner syncopes et MS (figure 1) (Goldenberg
et al., 2008). Les gènes responsables des formes les plus fré-
quentes, QTL1, QTL2, QTL5 et QTL6, codent pour des canaux
potassiques (KCNQ1, HERG = KCNH2, KCNE1, KCNE2 respecti-
vement) et celui responsable de la forme QTL3 code pour le
canal sodique cardiaque (SCN5A) (tableau 1). La prévalence
génétique est actuellement estimée à 1/2 000 individus. La bio-
logie moléculaire permet d’identifier une mutation dans 50 à
60 % des cas, mutation dont la transmission est le plus souvent
autosomique dominante. L’analyse de l’ensemble des tracés
ECG des membres d’une même famille améliore la sensibilité
et la spécificité pour un patient donné. Les anomalies de
durée et de morphologie de la repolarisation ventriculaire font
partie des critères diagnostiques du QTL. Le mode de déclen-
chement des événements rythmiques semble dépendre de la
forme génétique. En effet, le plus souvent, le facteur déclen-
chant d’une syncope ou d’un arrêt cardiaque en cas de QTL1
est un stress, surtout à l’effort. Les patients QTL2 ont, eux,
des syncopes ou des troubles du rythme survenant plutôt lors
d’une stimulation auditive ou à l’émotion, et les patients QTL3
Tableau 1.Les gènes impliqués dans les arythmies
ventriculaires primitives ou « canalopathies »
Syndromes Gènes Courant
QTL1 KCNQ1 Iks æ
QTL2 KCNH2 Ikr æ
QTL3 SCN5A Ina ä
QTL4 Ankyrine B
QTL5 KCNE1 Iks æ
QTL6 KCNE2 Ikr æ
Syndrome de Jervell KCNQ1 Iks æ
Syndrome de Jervell KCNE2 Ikr æ
Syndrome de Timothy CACNA1C Ca++ä
QT court KCNH2 Ikr ä
QT court KCNQ1 Iks ä
QT court KCNJ2 Ik1 ä
Syndrome de Brugada SCN5A Ina æ
TVC 1 RyR2 Ca ++ ä
TVC 2 CASQ2 Ca ++ ä
Syndrome d’Andersen KCNJ2 Ik1 æ
QTL : syndrome du QT long ; TVC : tachycardies ventriculaires caté-
cholergiques.
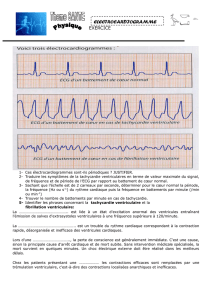
Figure 1. Tracé électrocardiographique en trois dérivations d’une jeune fille adressée pour syncope montrant des salves de torsades
de pointes. L’intervalle QT est très allongé (QTc = 500 ms).
Les canalopathies cardiaques
Épilepsies, vol. 22, n° 3, juillet-août-septembre 2010
231
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

durant le repos ou le sommeil (Schwartz et al., 2001). La sur-
venue des symptômes dépend de l’âge et du sexe. L’âge de sur-
venue du premier événement clinique est plus précoce chez les
garçons que chez les filles, mais, après l’âge de 15 ans, les fem-
mes sont plus symptomatiques que les hommes. Les syncopes
sont souvent associées à des convulsions qui sont parfois au
premier plan. Il reste très difficile d’évaluer le pronostic de
cette maladie pour un patient donné. L’incidence des événe-
ments cardiaques est plus importante dans les groupes QTL1
et QTL2 que dans le groupe QTL3, et augmente avec la valeur
de l’espace QTc, indépendamment du génotype. La mortalité
cardiaque est comparable dans les trois groupes, mais la létalité
des événements est plus élevée dans le groupe QTL3. Ainsi,
dans les formes QTLl et QTL2, les patients sont plus souvent
symptomatiques mais en meurent peu, alors que, dans la
forme QTL3, les patients sont moins symptomatiques, mais le
risque mortel à chaque syncope est plus important (Zareba et
Cygankiewcz, 2008)
Prise en charge thérapeutique
Le traitement de référence dans la prévention des syncopes
et de la MS sont les bêtabloquants, qui doivent être prescrits à
tous les patients symptomatiques. En effet, il y a une vingtaine
d’années, la mortalité rythmique spontanée était considérable :
de l’ordre de 70 % à 10 ans, survenant habituellement dans les
deux ou trois premières décennies. L’utilisation du traitement
bêtabloquant à partir des années 1975-1980 a permis de dimi-
nuer d’un facteur 10 cette mortalité. Dans notre expérience le
pourcentage de récidives sous traitement par nadolol, à une
posologie de 50 mg/m
2
, est nettement plus faible. Cette effica-
cité remarquable des bêtabloquants dans la prévention de la
MS est probablement à moduler en fonction du génotype.
L’effet bénéfique des bêtabloquants en monothérapie s’adresse
avant tout aux patients QTL1 et à certains patients QTL2 –
souvent associé à une stimulation cardiaque définitive –, mais
il est probablement peu adapté à certains patients QTL3, pour
lesquels un défibrillateur automatique implantable (DAI) est
plus indiqué. Ainsi, le traitement envisagé avant la connais-
sance du diagnostic génétique doit être rediscuté au vu du
génotype (Vincent et al., 2009). Dans tous les cas de QTL
confirmé ou supposé, des mesures de prévention doivent être
prises:
–dépistage et mise en route d’un traitement bêtabloquant chez
tous les sujets atteints, symptomatiques ou non, a fortiori chez
les enfants ;
–prévention des circonstances favorisant la survenue des TDP
(hypokaliémie, entre autres) ;
–contre-indication aux sports de compétition, hormis les
sports de classe 1A (Pelliccia et al., 2008), et pratique du sport
de loisir restreinte selon les recommandations (Maron et al.,
2004) ;
–remise d’une liste de médicaments contre-indiqués, car
connus pour allonger l’intervalle QT
1
.
Tachycardies ventriculaires catécholergiques
Les tachycardies ventriculaires catécholergiques sont carac-
térisées par des arythmies ventriculaires polymorphes de
déclenchement adrénergique. Elles surviennent essentielle-
ment chez des enfants et des adolescents, et sont responsables
de syncope et de MS, en l’absence de toute anomalie morpho-
logique cardiaque. Les accidents cardiologiques surviennent
dans un contexte émotionnel et/ou à l’effort, ou au cours de
noyade. Dans plus d’un tiers des cas, les syncopes à l’effort
s’accompagnent de convulsions, expliquant ainsi le retard au
diagnostic, car le traitement initial est souvent un traitement
antiépileptique initié par les neurologues. L’ECG initial en
dehors de tout épisode syncopal et de tout traitement montre
un intervalle QTc normal sur fond de rythme sinusal un peu
lent pour l’âge. Il est peu contributif. C’est l’épreuve d’effort,
qui est l’examen clé du diagnostic. Au cours de l’effort, il existe
un seuil d’apparition des troubles du rythme ventriculaire qui
est reproductible et spécifique à chaque patient. On observe
une progression caractéristique, avec des ESV isolées, mono-
morphes puis polymorphes, un bigéminisme, puis des salves
bidirectionnelles et, enfin, des salves ventriculaires polymor-
phes (figure 2). La biologie moléculaire trouve dans 70 % des
cas une mutation transmise de façon autosomique dominante
dans le gène codant pour le récepteur à la Ryanodine de type 2
(RyR2), intervenant dans la régulation du calcium cytoplas-
mique (Denjoy et al., 2005). La mortalité des TVC en l’absence
de traitement est très élevée : elle atteint 30 à 50 % à l’âge de
30 ans. De plus, il existe une corrélation entre l’âge de
survenue de la première syncope et la sévérité de la maladie,
avec un pronostic très péjoratif lorsque les pertes de connais-
sance surviennent très jeune. Les bêtabloquants réduisent
de façon significative les syncopes et la MS. Néanmoins, les
posologies de bêtabloquants utilisées dans la prévention des
syncopes pour les TVC sont élevées, souvent le double de celles
utilisées dans le SQTL. L’indication du DAI chez l’adolescent
avec TVC incomplètement contrôlée par une posologie élevée
de bêtabloquants doit être envisagée plus largement (Hayashi
et al., 2009). Compte tenu du risque de mort subite, qui peut
être inaugurale, il est recommandé de traiter par bêtabloquants
tous les sujets génétiquement identifiés et les sujets asympto-
matiques ayant des arythmies ventriculaires à l’effort. Toute
activité sportive sera contre-indiquée, y compris chez les
patients traités par bêtabloquants (Maron et al., 2004 ; Pelliccia
et al., 2008).
Syndrome de Brugada
Le syndrome de Brugada associe un retard de conduction
intraventriculaire droite et une anomalie de la repolarisation
sous la forme d’un sus-décalage du segment ST dans les précor-
diales droites (figure 3), pouvant se compliquer de fibrillation
ventriculaire et de MS. On estime la prévalence du syndrome
entre 4 et 12 % des morts subites, toutes étiologies confondues.
1
www.qtdrugs.org.
I. Denjoy, et al.
Épilepsies, vol. 22, n° 3, juillet-août-septembre 2010 232
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Le syndrome serait responsable de la moitié des morts subites
survenant chez des patients adultes avec un cœur apparem-
ment normal. L’aspect ECG est parfois intermittent et peut
être démasqué au cours d’un test pharmacologique par l’in-
jection d’un bloqueur de canaux sodiques (ajmaline ou flécaï-
nide chez l’enfant). Les sujets sont considérés comme atteints
en cas d’anomalies électrocardiographiques typiques (type 1),
détectées soit spontanément, sur l’ECG de surface, soit lors des
tests pharmacologiques de sensibilisation (Wilde et al., 2002).
Les symptômes, qui sont dans la moitié des cas des épisodes
de syncopes à l’emporte-pièce sans prodrome, surviennent
essentiellement chez l’homme de 40 ans. Les formes à révéla-
tion pédiatrique sont extrêmement rares et se manifestent le
plus souvent dans un contexte fébrile. Le seul traitement
ayant fait ses preuves dans la prévention de la MS est le défi-
brillateur automatique implantable (DAI) (Denjoy et al., 2007).
En présence d’un patient symptomatique (syncope, mort
subite récupérée) ayant un aspect ECG de Brugada de type 1,
un DAI sera proposé. La stimulation ventriculaire, dont la
rentabilité diagnostique et pronostique est discutée, peut être
Figure 2. Électrocardiogramme en 12 dérivations d’un enfant de 12 ans au décours d’un effort avec syncope et convulsions. Il existe
des extrasystoles ventriculaires polymorphes isolées et en salves rapides.
Figure 3. Électrocardiogramme en 12 dérivations enregistré chez un homme de 36 ans au décours d’une syncope montrant un aspect
de Brugada de type I dans les dérivations VI et V2, et un bloc auriculoventriculaire (BAV) du premier degré.
Les canalopathies cardiaques
Épilepsies, vol. 22, n° 3, juillet-août-septembre 2010
233
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

proposée. La pratique sportive de compétition est contre-
indiquée, hormis les sports de classe 1A. La pratique du sport
de loisir est possible, de façon restreinte et selon des recom-
mandations (Maron et al., 2004 ; Pellicia et al., 2008). La bio-
logie moléculaire trouve une mutation dans le gène SCN5A
codant pour le canal sodique cardiaque chez 20 % des sujets
atteints, et reste encore un outil de recherche.
Syndrome du QT court
Le syndrome du QT court, entité nouvellement décrite,
associe un intervalle QT court (QTc ≤300 ms), à un risque
élevé de syncope et MS par arythmie ventriculaire maligne
(Maury et al., 2008). Les syncopes et MS peuvent survenir au
repos mais également à l’effort. L’anomalie électrocardio-
graphique est parfois associée à de la fibrillation auriculaire de
survenue précoce. Sur le plan génétique, des mutations dans
les gènes HERG (= KCNH2),KCNQ1 et KCNJ2 ont été identifiées
chez des patients avec syndrome du QTC. L’exploration élec-
trophysiologique permet de documenter des périodes réfrac-
taires auriculaires et ventriculaires courtes et une vulnérabilité
ventriculaire chez la majorité des patients. Le seul traitement
àl’heure actuelle reste l’implantation d’un défibrillateur car-
diaque (Zareba et Cygankiewicz, 2008). Toute pratique sportive
est contre-indiquée.
Prise en charge familiale
Le dépistage cardiologique de l’un de ces syndromes repose
sur l’analyse de l’ECG, les antécédents personnels de chaque
membre de la famille, éventuellement l’échocardiographie ou
les tests de sensibilisation. Pour la plupart de ces syndromes,
le ou les gènes en cause sont connus et rendent possible, en
conjonction avec la clinique, le dépistage des apparentés pré-
symptomatiques ou symptomatiques dont le diagnostic
n’aurait pas été établi. Cette prise en charge ne peut être
considérée comme un examen de routine. Les indications
doivent être sélectionnées et sa pratique s’appuiera sur le
concours d’une équipe pluridisciplinaire constituée idéalement
d’un cardiologue, d’un généticien et d’un psychologue.
Le patient doit être informé des enjeux du test et des limites
du résultat quant à une éventuelle prise de décision. Un délai
de réflexion doit être respecté. L’annonce des résultats doit être
encadrée et le suivi programmé sur le long terme.
Conclusion
Les canalopathies cardiaques sont rares, mais elles exposent
les sujets qui en sont atteints à des syncopes et à la mort subite
par arythmie ventriculaire maligne. Le diagnostic impose une
prise en charge spécifique et modifie significativement le mode
de vie. L’origine génétique en fait une maladie familiale.
Les membres de la famille doivent être adressés à des centres
spécialisés dans ces pathologies pour une prise en charge par des
équipes multidisciplinaires associant un cardiologue, un géné-
ticien, un psychologue et une équipe de biologie moléculaire.
□
Conflit d’intérêt : aucun.
Références
Denjoy I, Postma A, Lupoglazoff J.M,et al. Tachycardies ventriculaires
catécholergiques chez l’enfant Arch Mal Coeur 2005 ; 98 : 506-12.
Denjoy I, Extramiana F, Lupoglazoff JM, Leenhardt A. Brugada
syndrome. Presse Med 2007 ; 36 : 1109-16.
Goldenberg I, Zareba W, Moss AJ. Long QT syndrome. Curr Probl
Cardiol 2008 ; 33 : 629-94.
Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of
arrhythmic events in catecholaminergic polymorphic ventricular tachy-
cardia. Circulation 2009 ; 119 : 2426-34.
Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommandations for
physical activity and recreational sports participation for young patients
with genetic cardiovascular diseases. Circulation 2004 ; 109 : 2807-16.
Maury P, Extramiana F, Sbragia O, et al. Short QT syndrome. Update
on a recent entity. Arch Cardiovasc Dis 2008 ; 101 : 779-86.
Pelliccia A, Zipes DP, Maron BJ. Bethseda Conference # 36 and the
European Society of cardiology Consensus Recommandations revisited. A
J Am Coll Cardiol 2008 ; 52 : 1990-6.
Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype phenotype corre-
lation in the long QT syndrome: specific triggers for life-threatening
arrhythmias. Circulation 2001 ; 103 : 89-95.
Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-
blockers in long-QT syndrome type 1: contribution of noncompliance
and QT-prolonging drugs to the occurrence of beta-blocker treatment
“failures”.Circulation 2009 ; 119 : 215-21.
Wilde A, Antzelevitvh C, Borggrefe M, et al. Proposed diagnosistic
criteria for Brugada syndrome : consensus report. Circulation 2002 ; 106 :
2514-9.
Zareba W, Cygankiewicz I. Long QT and short QT syndrome. Prog
Cardiovasc Dis 2008 ; 51 : 264-78.
En pratique
Les canalopathies sont une cause de syncopes avec
convulsions du sujet jeune peu fréquente, mais poten-
tiellement létale.
Les arythmies ventriculaires malignes (tachycardie
ventriculaire polymorphe, torsades de pointes, fibrillation
ventriculaire) sont souvent favorisées par la stimulation
adrénergique : sport, émotion, stress.
Le traitement consiste en :
–QTL et tachycardies ventriculaires catécholergiques :
bêtabloquants ;
–Brugada et syndrome du QT court : défibrillateur
automatique implantable ;
–pratique sportive restreinte selon les recommandations.
L’origine génétique est la pathologie familiale.
□
I. Denjoy, et al.
Épilepsies, vol. 22, n° 3, juillet-août-septembre 2010 234
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
1
/
5
100%