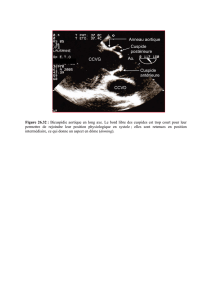
Génétique du rétrécissement aortique

Mini-revue
Sang Thrombose Vaisseaux 2010 ;
22, n° 5 : 257-63
doi: 10.1684/stv.2010.0492
Ge
´ne
´tique du re
´tre
´cissement aortique
Herve
´Le Marec
1,2,3
, Jean-Jacques Schott
1,2,3
, Vincent Probst
1,2,3
1
Inserm, UMR915, institut du thorax, CHU de Nantes, 44000 Nantes, France
2
CNRS, ERL3147, Nantes, 44000 France
3
Université de Nantes, Nantes, 44000 France
Résumé
.
Le re
´tre
´cissement aortique calcifie
´(RAC) est la maladie valvu-
laire la plus fre
´quente ; elle atteint 2 a
`5 % de la population a
ˆge
´e et repre
´-
sente la deuxie
`me indication de chirurgie cardiaque en France. On distin-
gue une forme conge
´nitale de RAC, caracte
´rise
´e par une malformation de
la valve dont la plus fre
´quente est la bicuspidie et une forme acquise ou
de
´ge
´ne
´rative qui a remplace
´la forme rhumatismale, devenue plus rare.
Avant 70 ans, la bicuspidie aortique est la plus fre
´quente, alors que la
forme de
´ge
´ne
´rative est pre
´ponde
´rante chez le patient plus a
ˆge
´. L’a
ˆge, le
sexe masculin, le taux de Lp(a), de LDL, l’hypertension arte
´rielle et le taba-
gisme sont des facteurs associe
´s au RAC. L’e
´tude de la ge
´ne
´tique du re
´tre
´-
cissement aortique n’a de
´bute
´que re
´cemment ; cependant, les donne
´es de
ge
´ne
´tique clinique et d’e
´pide
´miologie ge
´ne
´tique de
´ja
`disponibles montrent
que l’hypothe
`se ge
´ne
´tique n’est pas a
`ne
´gliger. Ainsi, le taux d’he
´ritabilite
´
de la bicuspidie est de pre
`s de 90 % et trois locus sur les chromosomes 18, 5
et 13 ont e
´te
´identifie
´s. L’intervention d’une mutation du ge
`ne NOTCH1 a
e
´galement e
´te
´de
´montre
´e ; ce ge
`ne intervient dans le de
´veloppement
embryonnaire des valves aortiques, et son alte
´ration semble favoriser la
bicuspidie aortique. Me
ˆme si de nombreux arguments laissent penser
qu’il existe une pre
´disposition a
`de
´velopper un RAC, il existe peu de preu-
ves du caracte
`re he
´re
´ditaire de cette affection. Des travaux re
´cents de notre
e
´quipe ont permis d’identifier des formes familiales de re
´tre
´cissement aor-
tique de
´ge
´ne
´ratif. Une liaison significative a e
´te
´mise en e
´vidence dans le
chromosome 16. Les donne
´es concernant la ge
´ne
´tique du re
´tre
´cissement
aortique restent encore limite
´es, il est cependant fort probable qu’il existe
une pre
´disposition ge
´ne
´tique a
`cette maladie. La de
´couverte du ro
ˆle de
NOTCH1 constitue une avance
´e majeure ; la constitution de grandes col-
lections d’ADN et de tissus de patients ope
´re
´s permettra certainement de
grandes avance
´es dans ce domaine.
Mots cle
´s:bicuspidie, calcification vasculaire, gène NOTCH1
Abstract
The Genetics of Aortic Stenosis
Calcified aortic stenosis (AS) is the commonest valvular disease : it affects
2 to 5% of the eldrely an dis the second indication for cardiac surgery
in France. There is a congenital form of calcified AS characterised by a
Tire
´sa
`part :
H. Le Marec
257
STV, vol. 22, n°5, mai 2010
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

valvular malformation, usually a bicuspid valve, and an acquired or dege-
nerative form which has replaced the rheumatic form which has become
less common.
Before the age of 70, bicuspid aortic valve is commoner whereas the dege-
nerative form is commoner in the more elderly patients.
Age, male gender Lp(a) and LDL cholesterol levels, hypertension and smo-
king are factors associated with calcified AS. The study of the genetics of AS
has only just begun: however, the available data of genetic clinical and epi-
demiological studies suggests that the genetic hypothesis cannot be neglec-
ted. The heritability of bicuspid aortic valves is nearly 90% and 3 loci on
chromosomes 18, 5 and 13 have been identified. The effect of a mutation
of the NOTCH1 gene has also been demonstrated: this gene intervenes in
the embryonic development of the aortic valve and an abnormality seems to
predispose to bicuspid aortic valves. Although many observations suggest
that there is a predisposition to calcified AS, there is little proof of the here-
ditary nature of this disease. Recent studies by our group have identified
familial forms of degenerative AS. A significant liaison has been demons-
trated on chromosome 16.
The data on the genetics of AS remains limited but a genetic predisposition
to this disease is very probable. The discovery of the role of NOTCH1 was a
major advance: the constitution of large banks of DNA and tissues of ope-
rated patients will certainly result in large advances in this field.
Key words:bicuspid valve, calcifid aortic stenosis, gene NOTCH1
Le rétrécissement aortique calcifié (RAC) est la
plus fréquente des maladies valvulaires, il tou-
che en effet 2 à 5 % de la population âgée. Il est
caractérisé par un épaississement et des calcifi-
cations des feuillets (sigmoïdes) et de l’anneau valvulaires,
responsables d’un obstacle à l’éjection du ventricule gauche.
Ses conséquences sont la création d’un gradient de pression
entre le ventricule et l’aorte initiale responsable d’une
hypertrophie progressive du ventricule gauche. C’est la
deuxième cause de chirurgie cardiaque en France, car il
n’existe pas d’autre solution thérapeutique que le remplace-
ment valvulaire dont l’indication devient indiscutable
quand surviennent les symptômes tels que l’insuffisance
cardiaque, les syncopes d’effort, voire l’angor. Le coût de
cette chirurgie est élevé et ses conséquences cliniques
potentiellement sévères chez des sujets âgés plus fragiles.
Enfin, les problèmes posés par cette pathologie risquent de
se majorer avec les avancées de la médecine qui ont pro-
longé considérablement l’espérance de vie.
Il existe deux grandes formes de RAC de l’adulte, une
forme congénitale caractérisée par une malformation de la
valve, dont la plus fréquente est la bicuspidie, et une forme
dite acquise ou « dégénérative » maintenant beaucoup plus
fréquente que la forme rhumatismale devenue rare.
Ces deux formes sont caractérisées par le développement
progressif d’un remodelage du tissu valvulaire qui va se
scléroser et se calcifier. Les valves bicuspides sont peu
sténosantes au début de la vie mais vont se calcifier plus
rapidement que les valves tricuspides normales. Ces bicus-
pidies seront responsables de RAC serrés survenant plus tôt
dans la vie. Ainsi, avant 70 ans, la bicuspidie aortique repré-
sente plus de 50 % des causes de rétrécissement aortique,
alors qu’après 70 ans, c’est la forme dite « dégénérative »
qui devient prépondérante [1].
Pendant de nombreuses années, le rétrécissement aortique a
été considéré comme un processus inéluctable, dégénératif,
lié au vieillissement et donc non modulable par une appro-
che préventive. La recherche de facteurs prédisposant à
cette affection n’était pas une préoccupation bien que des
études aient montré qu’un faible pourcentage (5 %) d’octo-
génaires étaient atteints de rétrécissement, modéré à sévère,
et que 14 % étaient porteurs de calcifications de la valve
aortique [2, 3], ce qui laisse supposer l’existence de facteurs
de prédisposition individuels qui ont été peu explorés.
Le caractère héréditaire de ces anomalies valvulaires a
donc été complètement ignoré, l’attention des cliniciens se
portant essentiellement sur les stratégies de prise en charge
des valvulopathies aortiques et plus particulièrement sur la
date de la chirurgie cardiaque.
Comme pour les dystrophies valvulaires myxoïdes, pour
lesquelles les bases génétiques deviennent maintenant évi-
dentes [4], il est probable que le rétrécissement aortique soit
258 STV, vol. 22, n°5, mai 2010
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

le résultat d’un processus lent de remodelage où un déter-
minisme génétique et de nombreux facteurs environnemen-
taux vont déclencher une succession d’événements qui vont
progressivement transformer une valve apparemment saine
en un appareil valvulaire profondément remanié (figure 1).
Le développement de thérapeutiques ciblées permettant de
ralentir ou d’éviter l’évolution vers un rétrécissement aor-
tique sévère devrait donc venir de l’identification des fac-
teurs initiaux du processus, des facteurs de risque et de la
compréhension des mécanismes moléculaires impliqués.
Ce n’est qu’à la fin des années 1990 et au début des années
2000 que les cliniciens et les chercheurs se sont penchés
avec plus d’attention sur la physiopathologie de ces attein-
tes valvulaires. La recherche s’est orientée dans deux gran-
des directions.
La première a consisté à étudier les facteurs cliniques et
biologiques associés au rétrécissement aortique et à recher-
cher, au niveau des valves pathologiques explantées lors de
la chirurgie cardiaque, la présence de modifications anato-
miques et moléculaires pouvant orienter vers un mécanisme
physiopathologique.
La seconde direction, plus récente, a été toute différente. Elle
est basée sur l’évidence que le rétrécissement aortique ne
touche qu’une faible proportion de sujets âgés, suggérant
l’existence d’une prédisposition individuelle, probablement
génétique, à développer un RAC. Elle consiste donc à recher-
cher des facteurs génétiques prédisposant à ces atteintes.
E
´tudes physiopathologiques
Dans une grande étude (Cardiovascular Health Study) [3],
l’âge, le sexe masculin, le taux de Lp(a), de LDL cholesté-
rol, l’hypertension et le tabagisme apparaissaient associés
au risque de rétrécissement aortique. L’analyse des valves
atteintes a conduit à la mise en évidence de nombreux pro-
cessus. Les contraintes mécaniques et le shear stress pour-
raient expliquer la survenue plus précoce des atteintes sur
valves bicuspides mais ne constituent pas en soi une cause
de rétrécissement aortique. Les dépôts lipidiques sous-
endothéliaux, l’accumulation de LDL oxydés, d’apolipo-
protéines, la présence de cellules spumeuses, de cellules
de l’inflammation, de cytokines pro-inflammatoires ont
conduit à évoquer des mécanismes similaires à ceux des
lésions athéroscléreuses [5]. D’autres études ont suggéré
le rôle du système rénine-angiotensine [6]. Enfin, beaucoup
d’éléments sont en faveur d’un processus cellulaire impli-
quant la transformation de fibroblastes de la valve aortique
en cellules ostéoblaste-like [7]. Actuellement, le rôle d’un
certain nombre de facteurs cliniques identifiés, tels que
l’hypercholestérolémie, n’a pas été confirmé par des études
d’intervention qui, quand elles ont été bien menées, se sont
révélées négatives [8]. De ces échecs, deux types de conclu-
sions peuvent être tirés : soit l’intervention est trop tardive
sur un remodelage moléculaire et tissulaire déjà très avancé,
soit les mécanismes identifiés ne sont que la conséquence
d’autres facteurs initiaux qui représentent probablement la
cible clé des interventions thérapeutiques mais qui restent
encore à identifier.
Approches de ge
´ne
´tique
dans le re
´tre
´cissement aortique
Plusieurs techniques de génétique sont possibles, chacune
ayant des avantages et des inconvénients : l’approche gène-
candidat, les études d’association et la génétique inverse.
Valve tricuspide
Valve bicuspide
Facteurs impliqués
Age
TGF β1
ApoE
VDR
Récepteur α œstrogène
Locus chr16
70500
NOTCH1
Locus
Chr18, 5, 13
Moléculaire :
Runx2/Cbfa1
Cellulaire :
Différenciation ostéoblastique,
calcifications,
dépôts lipidiques, inflammation
Facteurs de risque :
Shearstress, HTA,
syndrome métabolique
hypercholestérolémie,
diabète…
SténoseGènes Environnement Remodelage
Figure 1.Représentation schématique de l’évolution du rétrécissement aortique dit « dégénératif » et du rétrécissement aortique sur bis-
cupidie ainsi que des facteurs hypothétiquement liés à leur développement. Le rétrécissement aortique sur bicuspidie est caractérisé par
la présence d’un obstacle à l’éjection du ventricule gauche dès l’enfance, suivi d’un remaniement calcifié plus précoce. Les défauts géné-
tiques initiaux qui restent en partie hypothétiques vont être responsables, en conjonction avec les facteurs environnementaux, d’un remo-
delage moléculaire et cellulaire aboutissant au rétrécissement aortique calcifié.
259
STV, vol. 22, n°5, mai 2010
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Approche gène-candidat
L’approche gène-candidat est une approche risquée tout
particulièrement quand la physiopathologie n’est pas claire.
Elle consiste à rechercher des anomalies de séquence dans
des gènes choisis pour leur possible implication dans la
maladie. À côté du fait que cette approche ne permet pas
de vraies avancées dans la compréhension d’une maladie
(elle ne fait que confirmer des hypothèses déjà émises), ce
type de technique peut apporter de fausses conclusions, car
les polymorphismes, même rares, ne sont pas exception-
nels. Dans ce cadre, les résultats doivent obligatoirement
être complétés par une approche fonctionnelle, prouvant
l’implication du gène muté et, idéalement, par la démon-
stration de la liaison entre l’anomalie de séquence et la
ségrégation de la maladie dans une ou plusieurs familles.
Études d’association
Les études d’association visent à mettre en évidence, dans
la population, un déséquilibre de liaison entre un phénotype
et un polymorphisme. Elles exposent aux mêmes risques
que l’approche gène-candidat quand elles sont limitées à
l’étude d’un gène choisi pour sa potentielle implication
dans la physiopathologie de la maladie. Par ailleurs, cette
technique, qui nécessite une population témoin bien appa-
riée, est très dépendante de la taille de l’échantillon et de la
fréquence des polymorphismes dans la population étudiée.
Ce déséquilibre peut survenir par chance, ce qui rend abso-
lument nécessaire la réplication de l’étude sur plusieurs
cohortes. L’approche génome entier, rendue possible par
les nouvelles technologies de biologie moléculaire et l’uti-
lisation de puces SNP, est plus performante mais nécessite
de grandes cohortes. Dans tous les cas, le phénotype doit
être le plus précis possible, mais les approches de déséqui-
libre de liaison peuvent se révéler peu efficaces et nécessiter
de très grandes cohortes si la maladie est très hétérogène sur
le plan génétique, de nombreux gènes pouvant être respon-
sables d’un phénotype final commun.
Génétique inverse (linkage)
L’approche de loin la plus performante repose sur le linkage
et consiste à rechercher, dans de grandes familles sur plu-
sieurs générations, une liaison entre le phénotype et une
région du génome commune aux sujets atteints. Cette tech-
nique, comme les études GWAS, présente le grand intérêt
de faire abstraction de toute hypothèse physiopathologique
jusqu’àl’identification du locus. Ces deux approches,
GWAS et linkage, peuvent d’ailleurs être couplées et rendre
la démarche plus performante. Une fois le locus identifié, il
faudra séquencer les gènes à la recherche d’une anomalie de
séquence dont il faudra vérifier la ségrégation dans la ou les
familles, éliminer un polymorphisme rare, puis confirmer le
rôle fonctionnel de ces mutations. Cette technique a
l’inconvénient de nécessiter de grandes familles sur plu-
sieurs générations, ce qui n’est pas toujours réalisable, par-
ticulièrement quand le phénotype est d’apparition tardive.
C’est le cas du RAC qui apparaît chez des sujets âgés dont le
statut des ascendants est souvent difficile à retrouver et celui
de la descendance souvent indéterminé, les sujets étant trop
jeunes pour avoir développé la maladie. Enfin, si la péné-
trance est variable ou s’il existe de fréquentes phénocopies,
cette technique de génétique inverse peut être difficile à
mettre en œuvre.
La génétique du rétrécissement aortique n’a réellement
débuté qu’au cours de ces dernières années, et les connais-
sances sont encore parcellaires, mais les résultats déjà dis-
ponibles y compris les données de génétique clinique et
d’épidémiologie génétique montrent que l’hypothèse géné-
tique n’est pas à négliger et représente un enjeu majeur dans
la démarche physiopathologique.
Ge
´ne
´tique de la bicuspidie aortique
La bicuspidie aortique présente l’avantage d’être identi-
fiable dès les premières années de la vie et de pouvoir plus
facilement en tracer le caractère héréditaire. Dans les années
1990 et 2000, plusieurs travaux de recherche clinique ont
montré que l’origine de la biscuspidie aortique était essen-
tiellement génétique. Une fréquence anormale de biscupi-
die avait été retrouvée chez les apparentés au premier degré
de sujets atteints de cette affection et surtout une large étude
américaine a montré que le taux d’héritabilité de la biscus-
pidie était de près de 90 % [9]. Le même groupe, par une
approche d’analyse de liaison, a pu identifier trois locus sur
les chromosomes 18, 5 et 13 [10]. Ces travaux n’étaient pas
focalisés sur le RAC, mais essentiellement sur la bicuspidie.
Un pas important a été franchi quand le gène NOTCH1 apu
être mis en cause [11]. La voie de NOTCH1 a en effet été
identifiée par une démarche de linkage dans une famille
américaine d’origine espagnole, où la ségrégation d’une
pathologie aortique était certaine sur quatre générations.
Dix sujets étaient porteurs d’une atteinte valvulaire aortique
et, parmi ceux-ci, six étaient porteurs d’une biscuspidie et
trois avaient des valves calcifiées sans bicuspidie. Une ana-
lyse de liaison génétique a permis de localiser le gène en
cause dans une région de 9 cM (environ 3 Mbases) en
9q34-35 entre le marqueur D9S1826 et D9qter, avec un
LOD score significatif de 3,5 à 0 % de recombinaison.
Parmi les 30 gènes connus se trouvait NOTCH1, dont le
séquençage a permis d’identifier une mutation présente
chez tous les sujets atteints et absente chez les sujets sains.
Cette mutation R1108X est responsable d’un codon stop
prématuré qui doit se traduire par une protéine tronquée.
260 STV, vol. 22, n°5, mai 2010
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

L’étude d’une autre petite famille espagnole atteinte de
bicuspidie a permis d’identifier une délétion d’une base
(H1505del) dans ce même gène chez tous les sujets porteurs
de valves bicuspides. Cette seconde anomalie est respon-
sable d’un décalage du cadre de lecture dont les conséquen-
ces sont : 74 acides aminés incorrects suivis d’un codon
stop. Dans les deux cas, la protéine codée est probablement
non fonctionnelle, responsable d’une haplo-insuffisance.
Ces familles sont toutes deux atteintes de bicuspidies qui,
on le sait, favorisent le développement précoce d’un rétré-
cissement calcifié. Des mutations de NOTCH1 ont été
retrouvées dans deux autres cas sporadiques de bicuspidie.
La question qui se pose est de savoir si NOTCH1 est uni-
quement responsable de la biscuspidie ou si ce gène joue
aussi un rôle dans le développement de calcifications
comme le suggère la présence, dans la grande famille étu-
diée, de trois sujets atteints de RAC, apparemment sans
bicuspidie.
NOTCH1 code pour un récepteur de membrane probable-
ment impliqué dans l’embryogenèse cardiaque comme le
montre l’abondance des transcrits dans le mésenchyme de
la chambre de chasse qui va donner naissance aux valves
puis, après septation, dans les feuillets de la valve aortique
[12]. Par ailleurs, chez le poisson et la grenouille, NOTCH1
semble jouer un rôle important dans le développement des
valves [13]. Ces données plaident pour la responsabilité de
NOTCH1 dans la bicuspidie qui est supposée favoriser, par
les modifications hémodynamiques qu’elle induit, le déve-
loppement des calcifications.
Un des mécanismes proposés pour le développement des
calcifications passe par l’induction de la différenciation
des cellules valvulaires (fibroblastes et myoblastes) en
ostéoblastes. Dans les modèles animaux développant des
calcifications, un certain nombre de gènes sont up-régulés,
tels que ceux codant pour l’ostéocalcine, l’ostéopontine et
Runx, faisant évoquer l’implication de régulateurs de la
transcription de ces gènes dans le développement du
RAC. Dans les conditions normales, NOTCH1 active la
famille des Hrt (hairy-related transcriptional repressors)
qui inhibent l’activation Runx2, un activateur de l’expres-
sion d’ostéocalcine. Dans ces conditions, une mutation
perte de fonction de NOTCH1 qui est fortement exprimée
dans les valves pourrait favoriser le développement des cal-
cifications valvulaires via la baisse d’activation des Hrt
levant l’inhibition de Runx et favorisant l’expression de
gènes de la différenciation osseuse. Ainsi, la découverte
de NOTCH1 représente un pas important dans la compré-
hension des mécanismes conduisant au développement du
rétrécissement aortique. Il faut cependant noter qu’il semble
que les mutations de NOTCH1 soient rarement retrouvées y
compris dans les bicuspidies et que d’autres gènes sont pro-
bablement impliqués. Les familles de gènes jouant un rôle
dans la différenciation ostéogénique et fortement exprimés
dans les valves cardiaques représentent cependant une piste
privilégiée pour la recherche de facteurs génétiques prédis-
posant au RAC.
Ge
´ne
´tique du re
´tre
´cissement aortique
calcifie
´dit « de
´ge
´ne
´ratif »
Le rétrécissement aortique « dégénératif » qui survient sur
des valves qui initialement semblaient normales est carac-
térisé par une expression tardive dans la vie, généralement
après la soixantaine. Cela rend son étude génétique beau-
coup plus difficile, car les techniques de génétique inverse
sont difficilement utilisables dans des pedigrees où une
seule génération apparaît atteinte. Il est, en effet, rarement
possible de disposer connaître le phénotype et de disposer
de l’ADN des parents de patients atteints de rétrécissement
aortique dégénératif. Le challenge que représente l’identifi-
cation des facteurs génétiques de cette maladie peut être
abordé de différentes manières mais dans tous les cas néces-
site la mise en place d’efforts considérables pouvant asso-
cier l’épidémiologie génétique, la recherche de gènes
candidats ou les études d’association.
L’e
´pide
´miologie ge
´ne
´tique
Même si de nombreux arguments cliniques font penser
qu’il existe une prédisposition à développer un RAC, il
existe actuellement peu de preuves du caractère héréditaire
de cette affection. Les premiers éléments de preuve ont été
apportés par un travail d’épidémiologie génétique qui a été
débuté au début des années 2000 par notre équipe [14].
La région de Nantes a pour particularité d’avoir un seul
centre de chirurgie cardiaque et un taux de fuite extrême-
ment limité, ce qui fait que depuis plusieurs décennies, tous
les rétrécissements aortiques diagnostiqués dans notre
région et nécessitant un geste chirurgical sont opérés dans
le service de chirurgie cardiaque du CHU de Nantes. Une
deuxième particularité de notre région est la très faible
mobilité de sa population. En effet, en dehors des grandes
villes, l’étude généalogique de la population montre que
pour un individu né dans une commune rurale, les ancêtres,
sur six générations, sont retrouvés à plus de 90 % dans un
rayon de 10 km. En tenant compte de ces caractéristiques,
nous avons rapporté le nombre de cas opérés sur dix ans à la
population existant dans leurs communes d’origine à la
période de leur naissance. Il s’agit bien sûr d’une évaluation
approximative de la prévalence du rétrécissement aortique
dégénératif sévère dans les différentes communes de notre
région, mais les résultats obtenus ont été suffisamment
clairs pour orienter notre recherche génétique. En effet, le
261
STV, vol. 22, n°5, mai 2010
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
 6
7
6
7
1
/
7
100%