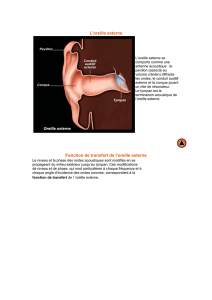
Aplasie de l'oreille : Aperçu de l'aplasie majeure

DOSSIER
25
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - n° 246 - octobre 1999
DE QUOI PARLONS-NOUS ?
Une aplasie est un défaut de développement d’un tissu qui a
gardé une forme embryonnaire.
En France, nous distinguons, suivant M. Ombredanne, d’une
part l’aplasie majeure (AM), constituée de quelques reliquats
de pavillon, d’une absence de conduit et de malformations
ossiculaires et, d’autre part, l’aplasie mineure (Am), consti-
tuée d’un pavillon et d’un conduit auditif externe normaux ou
subnormaux et de malformations ossiculaires. L’aplasie
majeure pose les problèmes plastique du pavillon et fonction-
nel auditif du conduit auditif externe et de l’oreille moyenne.
L’aplasie mineure pose essentiellement le problème fonction-
nel de l’oreille moyenne.
Dans ces deux formes d’aplasie, l’oreille interne est sous-
entendue comme étant normale ; ce sous-entendu correspond à
une réalité embryonnaire et pathologique, les oreilles externe
et moyenne dérivant d’éléments branchiaux, alors que l’oreille
interne dérive de la capsule otique ; l’aplasie de l’oreille est
essentiellement une pathologie d’origine branchiale.
Les Anglo-Saxons, dans leurs articles à finalité plastique, utili-
sent préférentiellement le terme de microtie, qui concerne le
pavillon et, dans leurs articles à finalité fonctionnelle, le terme
d’atrésie du conduit. Si le terme de microtie est clair, le terme
d’atrésie, qui correspond à un développement incomplet d’un
conduit avec ses multiples variantes, peut ainsi évoquer pour
l’ORL l’atrésie de l’œsophage, qui comporte une solution de
continuité de l’axe œsophagien avec un segment inférieur
indépendant du segment supérieur. Or, cette forme particulière
d’atrésie n’existe pas au niveau du conduit ; en effet, pour des
raisons embryologiques, le recouvrement épithélial du fond du
conduit et de la face externe du tympan ne peut être que d’ori-
gine ectodermique, provenant de la surface de l’embryon ; en
d’autres termes, en dedans d’un cul-de-sac d’un conduit bien
fermé, il n’y a pas de danger de trouver un autre segment de
conduit épithélialisé avec ses complications potentielles.
Exemples :
– un conduit en entonnoir, même très serré, est une Am car il
existe une continuité épithéliale entre l’orifice externe du
conduit et la membrane tympanique normale ou petite ; il y a
un risque de rétention épithéliale et sa canaloplastie de simple
agrandissement est relativement facile ;
– le conduit de l’AM est absent ou s’arrête totalement au bout de
quelques millimètres ; il n’y a pas de risque de rétention épithé-
liale et sa canaloplastie de création d’un néocanal est difficile.
En pratique :
– pour rapporter de grandes populations et du fait de la propor-
tionnalité quasi constante entre le degré des malformations du
pavillon, du conduit et de l’oreille moyenne, il est indifférent
d’utiliser les termes d’aplasie majeure, de microtie, ou d’atré-
sie du conduit ;
– pour décrire un patient donné, du fait de l’existence rare
mais possible de pavillons normaux sans conduit (AM) ou
d’anoties (absence de pavillon) avec conduits (Am), le prati-
cien est condamné à décrire pour chaque patient ces trois élé-
ments suivant les données qu’il possède : cliniques, audiomé-
triques, radiologiques voire opératoires.
Nous ne parlerons ici que des aplasies majeures. Bien que de
diagnostic facile, sa fréquence est peu précise, environ un cas
pour 5 000 à 10 000 naissances.
L’HYPOTHÈQUE VENTILATOIRE
L’aplasie majeure d’oreille s’accompagne toujours d’un cer-
tain degré d’hypoplasie mandibulaire homolatérale. Cette der-
nière peut induire une gêne ventilatoire obstructive ou une dif-
ficulté d’intubation éventuellement nécessaire à un examen
complémentaire par exemple. L’examen de la cavité buccale à
l’abaisse-langue et l’observation de la qualité du sommeil suf-
fisent à lever cette rare mais importante hypothèque.
L’AUDITION
L’AM, avec son absence de conduit, de membrane tympanique
et la présence de malformations ossiculaires, induit un déficit
auditif transmissionnel de 70 dB.
– Dans 80 % des cas, l’AM est unilatérale.
– Dans 15 % des cas, elle est bilatérale et alors, le plus sou-
vent, dans le cadre d’un syndrome (cf. infra).
– Dans 5 % des cas, elle est associée à une aplasie mineure
controlatérale qui risque d’être méconnue.
Dès lors, face à une AM d’apparence unilatérale, il est évident
que les possibilités auditives de l’enfant dépendront de
l’oreille controlatérale dont il faut connaître la valeur fonction-
nelle dès les premières semaines de vie. L’enregistrement des
oto-émissions provoquées (OEP) de cette oreille apparemment
saine se révèle remarquablement performant ; l’anomalie ou
l’impossibilité de la réalisation de cet examen doit conduire à
l’étude des potentiels évoqués auditifs du tronc cérébral
(PEATC) avant 6 mois.
L’aplasie de l’oreille
●Y. Manac’h*
* Service ORL, hôpital Necker-Enfants Malades, 149, rue de Sèvres, 75015 Paris.

●
●Première situation (80 % des cas) : l’AM est unilatérale
et l’oreille controlatérale saine.
La constatation de la normalité de l’audition de l’oreille
controlatérale permet d’être rassuré quant à la possibilité de
l’enfant pour une acquisition du langage et une scolarité nor-
males. Il faut reconnaître que dans la période néonatale, cette
tranquillité acquise par le médecin n’est pas toujours comprise
par la famille. On peut alors s’aider de quelques phrases : “il
suffit d’une bonne oreille pour bien entendre...”, “il y a des
enfants qui naissent sans malformation évidente et chez qui on
découvre, vers 5-6 ans, à l’âge du téléphone, une surdité bien
plus importante, unilatérale, qui, jusque-là, était méconnue, car
sans gêne...”, “si une surdité unilatérale acquise peut être
gênante, une surdité congénitale l’est beaucoup moins...”, “en
outre, si votre enfant entend mal du côté de sa malformation, la
partie noble de l’audition, l’oreille interne, est présente ;
l’audition de cette oreille malformée est donc en réserve ; il
suffirait par exemple d’un appareillage pour que, si un malheur
survenait du côté de sa bonne oreille, il puisse entendre norma-
lement...”. Cette oreille “unique” nécessitera cependant, par
prudence, un suivi otoscopique et au moins acoumétrique
annuel. Le premier contrôle ORL est ainsi effectué vers l’âge
de 12-14 mois pour vérifier la mise en place normale du lan-
gage. Les parents sont alors rassurés.
Deux questions peuvent alors se poser :
– Le bilan radiotomodensitométrique (TDM), son indication et
sa date : il faut reconnaître que, dans ce cadre clinique, la
TDM apporte peu de choses : du côté de l’AM, il n’y a pas de
risque d’inclusion épithéliale (cf. supra) ; il n’y a pas d’indica-
tion de chirurgie à but fonctionnel (cf. infra) ; dès 5-6 ans, on
aura une certitude audiométrique de la normalité de l’oreille
interne du côté aplasié. Cependant, il est classique de deman-
der un bilan TDM, mais il doit être de bonne qualité et non pas
constitué de quelques coupes axiales chez un enfant qui a
bougé avec de mauvaises reconstructions coronales ou sagit-
tales. Il semble préférable d’attendre l’âge de 6-7 ans, âge
auquel la sagesse de l’enfant offre une immobilité suffisante
sans besoin de prémédication ni d’anesthésie générale pour
avoir de bonnes images coronales et axiales natives.
– Les otites sur AM. Aucune publication ne fait état d’otite
moyenne aiguë sur AM et, a fortiori, de complication. Les acci-
dents infectieux observés résultent d’une malformation du
pavillon avec, par exemple, une conque rétentive, ou surviennent
sur une fausse AM qui était en fait une Am avec un conduit en
sablier conduisant à un accident otitique externe. Il est donc
important de faire cette différence entre AM et Am ; dans les
rares cas où l’examen clinique ne le permet pas, le bilan TDM
permet de trancher. Pour expliquer cette absence d’accidents oti-
tiques aigus de l’oreille moyenne qui doivent pourtant exister, on
est condamné à se rappeler que la majorité des otites moyennes
aiguës se guérissent spontanément ou bien qu’il s’agit le plus
souvent d’une affection bilatérale, alors diagnostiquée par l’exa-
men du côté non malformé, et qui bénéficie d’un traitement par
voie générale à efficacité bilatérale. Acceptons le fait que l’otite
moyenne aiguë purulente sur AM n’est pas un problème.
La famille et nous-même aimerions quand même normaliser
l’audition de cette AM ! Voyons les résultats de cette chirurgie
et éliminons les “star patients” que l’on peut retrouver dans la
littérature, mais qui sont rares et imprévisibles. Sur des patients
sélectionnés (dont le bilan TDM montre une belle cavité
d’oreille moyenne et un étrier présent...), le gain auditif postopé-
ratoire moyen est de 20 dB pour une perte originelle de 70 dB.
Où est la stéréoacousie recherchée ? Il semble actuellement plus
raisonnable de renoncer à cette chirurgie en attendant qu’elle
fasse de réels progrès, et ce d’autant plus qu’elle est pénalisante
sur le plan plastique. De même, lors de la reconstruction plas-
tique du pavillon, il faudra veiller à ce que la technique
employée ne gêne pas notre successeur otologiste (cf. infra).
L’appareillage de type CROSS avec un micro du côté de l’AM
et un embout ouvert du côté de l’oreille normale est en règle
refusé par les patients, même après essai. Sauf progrès chirurgi-
cal, cet enfant ne sera donc ni policier, ni militaire... mais il
pourra être, sans chirurgie ni aide auditive, comme les autres,
premier en classe... cela paraît être le principal !
●
● Deuxième situation (15 % des cas) : l’AM est bilatérale.
Nous avons un déficit auditif bilatéral de 70 dB incompatible
avec une normale acquisition spontanée du langage. L’examen
audiométrique subjectif adapté à l’âge et les PEATC sont obli-
gatoires. Le nourrisson est appareillé par voie osseuse sur
serre-tête avec une prise en charge orthophonique dès qu’il se
tient assis, vers 6 mois. Cette situation est beaucoup plus
lourde pour la famille et l’enfant mais, avec ces contraintes,
l’acquisition du langage et une scolarité quasi normale sont
préservées. Vers 5-7 ans, l’appareillage par voie osseuse
devient psychologiquement pesant : l’appareillage est visible,
le serre-tête se mobilise lors des activités physiques et, malgré
l’appareillage, l’enfant peut être gêné pour l’apprentissage des
langues étrangères.
On peut alors proposer un appareillage en conduction osseuse
directe sur vis de titane (BAHA), ou tenter une chirurgie fonc-
tionnelle qui est devenue, grâce à quelques progrès, acceptable
ici, dans le cadre d’une AM bilatérale qui nécessite a priori de
toute façon un appareillage auditif. Cette chirurgie a toujours
des gains auditifs en règle insuffisants, nous l’avons dit. Elle
conduit souvent à un néoconduit médio-conqual avec trou noir
dysharmonieux ; cependant, l’utilisation d’un lambeau galéal
pour venir tapisser l’ensemble du néoconduit (dont le tympan)
et qui sert de sous-sol bien vivant à son recouvrement épithé-
lial par une greffe de peau fine permet d’obtenir, dans plus de
70 % des cas, ce qui est acceptable, un néoconduit sec et
stable, appareillable par voie aérienne, en intra-conqual ou
intra-conduit (figure 1). En d’autres termes, si le problème de
la création d’un néoconduit est solutionné, même en cas de
base pétreuse très pneumatisée, le problème columellaire – par
exemple en cas d’ectopie du nerf facial gênant l’accès à la
fosse ovale, si on ne veut pas prendre le risque, pour le court et
le long terme, d’une fenestration labyrinthique – reste entier. Il
y a bien sûr des “star patients” sans nécessité d’appareillage
après chirurgie fonctionnelle, mais il y a aussi ceux dont le
gain auditif est nul et dont l’appareillage par voie aérienne est
moins performant que l’appareillage par voie osseuse. Pour
être dans la situation d’un appareillage par voie aérienne per-
formant, il faut en pratique un gain postopératoire de 20 dB, et
nous avons vu que cela est réaliste. Cette chirurgie à but fonc-
tionnel ne peut se faire qu’avec une parfaite connaissance
audiométrique et radiologique et l’assurance d’une bonne col-
DOSSIER
26
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - n° 246 - octobre 1999

laboration de l’enfant ; elle est réalisée au cours du deuxième
temps de reconstruction du pavillon vers 7-9 ans ; en cas
d’échec, l’utilisation d’un BAHA reste possible.
Le BAHA est une remarquable alternative à l’appareillage
auditif par voie osseuse sur serre-tête. Nécessitant une corti-
cale temporale d’au moins 3 mm d’épaisseur et un peu d’atten-
tion de la part de son porteur, car il reste fragile, il n’est en
pratique mis en place que vers 7-8 ans. Imposant définitive-
ment le port d’un pilier traversant le scalp et d’un petit boîtier
crânien que le patient cherche à cacher sous sa chevelure, il est
donc logique de proposer dans un premier temps une chirurgie
fonctionnelle. Si le BAHA est mis en place en première inten-
tion, on se doit de le placer sur une zone non gênante vis-à-vis
de toute chirurgie plastique ou fonctionnelle ultérieure.
●
●Troisième situation (5 % des cas) : l’AM est associée à
une Am controlatérale.
L’Am doit être appareillée par voie aérienne. Si la forme du
pavillon ou du conduit ne le permet pas, ce peut être l’indication
d’un appareillage par voie osseuse sur serre-tête, temporaire,
avant que ne soit réalisée une canaloplastie ou une otoplastie
permettant cet appareillage. La canaloplastie de simple agrandis-
sement, qui ne touche pas l’oreille moyenne, est tout à fait réali-
sable chez le petit enfant. La pratique d’une tympanoplastie du
côté de l’aplasie mineure nécessite une parfaite connaissance
radiologique et fonctionnelle (dont une audiométrie subjective)
des deux oreilles, ce qui la rend difficilement réalisable chez le
petit enfant avant 5-6 ans. Par ailleurs, seule l’exploration chi-
rurgicale permet de dire avec certitude quel est l’élément de la
chaîne ossiculaire à corriger, et ce peut être l’étrier ; du fait du
risque statistiquement faible, mais inhérent à toute chirurgie sta-
pédienne, le contrat préopératoire doit être clair : soit faire une
simple tympanotomie exploratrice si le problème se révèle sta-
pédien, soit avoir évoqué le petit risque (en fréquence) d’un
appareillage par voie osseuse sur l’AM, soit encore débuter par
une chirurgie fonctionnelle du côté de l’AM. La tympanoplastie
sur Am permet de normaliser l’audition sur le long terme chez
50 % des opérés et ses meilleurs résultats sont obtenus en cas de
correction d’une anomalie stapédienne. Tels sont les arguments
qui participent actuellement à la décision thérapeutique.
LE PAVILLON
Dans le cadre des aplasies d’oreille, toutes les malformations
sont observables, depuis le pavillon normal jusqu’à l’anotie en
passant par tous les degrés de pavillon en cornet. La forme de
loin la plus fréquente est la microtie (figure 2),constituée d’un
lobule verticalisé surmonté d’un reliquat cartilagineux. Si le but
du chirurgien est d’apporter une satisfaction postopératoire au
patient, on peut dire que le problème est potentiellement solu-
tionné. Si son but est de construire une oreille normale, il y a
encore des progrès à faire, surtout dans la régularité des résultats.
Trois possibilités de prise en charge existent :
– L’abstention thérapeutique. Elle est rarement exprimée par
les enfants : la phrase “Docteur, laisse moi-ma petite oreille
cassée” demeure exceptionnelle.
– La mise en place d’une épithèse sur des vis de titane : sa
mise en place est chirurgicalement simple, son résultat plas-
tique dépend de l’habilité de l’épithésiste. Le pavillon de sili-
cone paraît normal à 2 m, mais, de plus près, on peut se rendre
compte qu’il s’agit d’une épithèse comme on peut reconnaître
une main de matière plastique. Elle impose en règle l’ablation
des reliquats de pavillon afin d’avoir une bonne continuité
entre le plan jugal et le bord antérieur de l’épithèse. Elle est
amovible, se fixant par des clips sur des piliers transcutanés
qui prolongent les vis intra-osseuses, et elle se retire le soir.
Elle s’altère avec le temps et doit être refaite au bout de 2 à
4ans. S’il s’agit d’une méthode thérapeutique tout à fait inté-
ressante chez les adultes, en particulier “mariés”, son caractère
27
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - n° 246 - octobre 1999
Figure 1. Microtie
après reconstruc-
tions plastique et
fonctionnelle. Le
néoconduit nuit au
résultat plastique
mais permet un
appareillage par
voie aérienne en
intraconduit.
NB : le sillon rétro-
auriculaire, en par-
tie comblé par la
cale cartilagineuse
d’élévation, ne per-
met pas, en règle,
un appareillage en
contour d’oreille.
Figure 2. Microtie
typique avec son
lobule verticalisé
surmonté d’un
petit reliquat
cartilagineux.

amovible fait qu’elle est en règle générale mal acceptée chez
l’enfant et l’adolescent. Elle coupe les ponts pour une inter-
vention de reconstruction d’un néo-pavillon, du moins dans sa
technique habituelle.
– Une reconstruction d’un pavillon à l’aide d’une charpente car-
tilagineuse construite à partir d’une autogreffe de cartilage cos-
tal. C’est actuellement la méthode la plus satisfaisante, réalisée
suivant la technique de S. Nagata. Son résultat est correct, don-
nant satisfaction au patient, sans être parfait. C’est une oreille
“bien au patient” permettant de mener une vie normale
(figure 3). Sa réalisation nécessite deux conditions : la première
est le poids de l’enfant, qui doit être supérieur ou égal à 25 kg
pour avoir un volume costal suffisant ; l’intervention est donc
rarement réalisée avant 7-8 ans ; la deuxième, comme dans toute
chirurgie plastique, est la nécessaire demande de l’enfant et non
pas celle de la famille ; l’enfant doit avoir compris le résultat
visé et la lourdeur au moins de son premier temps opératoire qui
entraîne, pendant quelques jours, un réel inconfort lié au prélè-
vement costal, comparable à celui occasionné par une fracture
de côte malgré le traitement antalgique. En pratique, la demande
et la collaboration potentielle de l’enfant se lisent dans son
regard. Le premier temps opératoire consiste, après avoir retiré
les éventuels reliquats cartilagineux de la microtie, à mettre la
charpente du nouveau pavillon sous la peau auriculaire et à
transposer en bonne place le lobule. Le deuxième temps, plus
léger, consiste à élever le néo-pavillon, c’est-à-dire à lui donner
du relief par rapport au plan de la face en utilisant une cale carti-
lagineuse glissée derrière le pavillon, cale précédemment laissée
sous la peau thoracique lors du premier temps opératoire. C’est
à ce niveau que le chirurgien conscient du problème auditif, qui
sera potentiellement traité dans l’avenir, doit légèrement modi-
fier la technique originelle en utilisant un lambeau de tissu péri-
osté de la base mastoïdienne ou un lambeau galéal occipital pour
recouvrir cette cale, et non pas le lambeau de galéa temporal de
la technique princeps. Un troisième temps d’éventuelles
retouches peut être nécessaire.
La technique de S. Nagata actuellement utilisée résulte des
progrès apportés successivement par M. Ombredanne, E. Pon-
cet, R. Tanzer et B. Brent. Son principal inconvénient demeure
la nécessité d’un important prélèvement cartilagineux costal
par thoracotomie ; les prochains progrès devraient venir de
l’utilisation d’une charpente issue d’une culture d’une simple
biopsie de cartilage autogène ; ce type de culture est dès à pré-
sent réalisable, mais il ne permet d’obtenir qu’une lame tissu-
laire plane qu’il n’est pas encore possible de modeler en un
volume stable dans le temps.
LES SYNDROMES ET LA GÉNÉTIQUE
Face à une AM de l’oreille, la recherche d’un syndrome est
systématique, d’une part pour connaître les éventuelles malfor-
mations associées et établir un programme thérapeutique pré-
coce et global et, d’autre part, pour permettre d’orienter le
conseil génétique, parce qu’un grand nombre de ces syn-
dromes est d’origine génétique. Les familles en sont toujours
demandeuses, que ce soit pour de futurs collatéraux ou des
descendants ; le moment d’en parler est une question de doigté
au cours du suivi du patient et de sa famille. Pour établir le
diagnostic syndromique, il est pratique de distinguer :
1. Les dysmorphoses sévères du pavillon de type microtie
Elles conduisent à rechercher d’autres anomalies essentielle-
ment locales ou régionales au niveau des autres dérivés des
arcs branchiaux. L’oreille interne est donc en règle indemne.
●
● Nous avons vu l’aplasie majeure avec ses formes unilaté-
rales, plus rarement bilatérales, ou associée à une aplasie
mineure controlatérale. Elle s’accompagne quasi constamment
d’au moins une note d’hypoplasie mandibulaire homolatérale,
ce qui devrait lui permettre de s’appeler “syndrome oto-mandi-
bulaire”. La prise en charge de cette hypoplasie mandibulaire
dépend de son retentissement esthétique facial et fonctionnel
au niveau de l’articulé dentaire ; elle est orthodontique ou chi-
rurgicale chez le grand enfant ; l’ostéodistraction mandibulaire
est en cours d’évaluation de ce cadre. Sa fréquence est d’envi-
ron 1 à 2 cas pour 10 000 naissances. L’interrogatoire retrouve
exceptionnellement le rôle d’un agent tératogène qui, s’il est
retrouvé, induit préférentiellement une malformation bilaté-
rale. Les antécédents familiaux sont en règle générale absents
(3 % des cas). Sur une population de 500 syndromes oto-man-
dibulaires, isolés, unilatéraux et sans antécédent familial sui-
vis dans le service, nous avons pu établir un risque de récur-
rence de 2,7 pour 1 000 tant pour les futurs collatéraux que
pour la descendance. Ce chiffre est très utile aux familles.
●
● Le syndrome de Goldenhar. C’est une dysmorphose
sévère du pavillon qui s’intègre dans un ensemble malformatif
avec des atteintes oculaires et vertébrales. Ce syndrome est
également appelé syndrome oculo-auriculo-vertébral ou micro-
somie hémifaciale. La dysmorphose du pavillon est variable,
allant d’un pavillon de structure anarchique à l’anotie. Cette
dysmorphose du pavillon est souvent associée à des fibro-
enchondromes préauriculaires ou siégeant sur une ligne allant
de la commissure buccale à l’oreille. L’atteinte peut être bila-
térale (30 % des cas), mais toujours franchement asymétrique
DOSSIER
28
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - n° 246 - octobre 1999
Figure 3. Microtie
après reconstruc-
tion plastique sui-
vant la technique
de S. Nagata.

(figures 4 et 5). L’hypoplasie mandibulaire est plus marquée
que dans le syndrome oto-mandibulaire. Au niveau régional,
on trouve une atteinte ophtalmologique quasi constante sous la
forme d’une tuméfaction épibulbaire, plus rarement d’une ano-
malie de la fente palpébrale, d’un colobome de la paupière
supérieure, voire d’une microphtalmie. D’autres anomalies
faciales peuvent être retrouvées, comme une paralysie faciale,
une fente vélo-palatine, une macrostomie. Le rachis présente
une anomalie dans 30 % des cas, sous la forme d’une fusion
vertébrale ou d’une hémi-vertèbre, habituellement stable. Plus
rares, mais à rechercher systématiquement, sont les anomalies
cardiaques et rénales. Le système nerveux central présente
dans 5 à 15 % des cas des anomalies radiologiques. L’intelli-
gence est en règle normale. Sur le plan thérapeutique spéci-
fique, la tuméfaction épibulbaire peut nécessiter une prise en
charge chirurgicale pour des raisons esthétiques ou fonction-
nelles. Comme pour le syndrome de Treacher Collins que nous
verrons ci-après, nous sommes ici dans le cadre d’une atteinte
chronologiquement prébranchiale d’une neurocristopathie
rhombencéphalique. L’incidence du Goldenhar est de 1 pour
5000 naissances mais ce chiffre, issu de la littérature, paraît
élevé. Il est dans la majorité des cas sporadique et d’étiologie
inconnue ; cependant, 1 ou 2 % de formes familiales, d’expres-
sion intrafamiliale très variable, peuvent suggérer dans ces cas
une transmission autosomique dominante. Le risque de récur-
rence est faible et évalué à environ 5 %.
●
● Le syndrome de Treacher Collins. La dysmorphose du
pavillon n’est pas l’élément le plus constant de ce syndrome
mais elle est, quand elle existe, le signe d’appel le plus voyant.
L’atteinte faciale, toujours bilatérale, est symétrique. Elle
donne au visage une forme de “tête d’oiseau” qui oriente le
diagnostic (figure 6). Ce syndrome est encore appelé syn-
drome de Franceschetti-Zwahlen-Klein ou dysostose mandi-
bulo-faciale. La dysmorphose du pavillon est donc très
variable. Le pavillon est parfois normal, mais, dans 60 % des
cas, il s’agit d’une microtie. L’hypoplasie mandibulaire com-
porte une ouverture de l’angle de la mâchoire et une
rétrogénie ; surtout au niveau du tiers moyen de la face, il
existe une hypoplasie malaire (80 % des cas), avec des pom-
mettes effacées qui induisent une obliquité anti-mongoloïde
des fentes palpébrales. Dans ce cadre régional il peut exister
un colobome des paupières inférieures (70 % des cas), voire de
l’iris. Une fente vélo-palatine est présente dans 35 % des cas.
L’intelligence est en règle normale. Sur le plan thérapeutique
spécifique, la dysmorphose du tiers moyen de la face est prise
en charge parallèlement à l’hypoplasie mandibulaire en utili-
sant des greffes d’apposition osseuse pour le malaire avec par-
29
La Lettre d’Oto-rhino-laryngologie et de chirurgie cervico-faciale - n° 246 - octobre 1999
Figure 4.
Syndrome de
Goldenhar avec
son atteinte sou-
vent bilatérale mais
asymétrique ; ici,
microtie d’un côté
et pavillon en
cornet de l’autre.
Figure 5. Le même
cas que celui de la
figure 4 pour mon-
trer le kyste épibul-
baire et le colobome
irien supéro-
externe.
Figure 6.
Syndrome de
Treacher Collins
(ou de
Franceschetti)
avec son hypoplasie
malaire, son colo-
bome palpébral
inféro-externe et
son hypoplasie
mandibulaire. La
malformation est
symétrique. Ce cas
a été choisi pour
montrer que la
microtie n’est pas
constante dans ce
syndrome.
 6
6
1
/
6
100%