Apoptose et maladies neurodégénératives

apoptose est un processus indispensable à l’homéo-
stasie tissulaire des métazoaires. Elle intervient dans la
suppression de cellules en surplus, endommagées ou
âgées, dans les tissus sains tout comme lors de la maturation des
organes, au cours de l’embryogenèse, notamment lors de la neu-
rogenèse et de la fabrication de la mémoire immunitaire (1).
Au cours du développement du système nerveux, l’apoptose
s’observe dès la formation du tube neural et persiste jusqu’à
la différenciation du réseau neuronal final. Plus de 50 %
des neurones seraient perdus par ce processus, au cours du
développement.
Lors du vieillissement d’un individu, il arrive parfois que l’on
observe des dérégulations de l’apoptose au niveau du système
nerveux central (SNC), comme lors des maladies neurodégé-
nératives (MND), où elle intervient de façon excessive. C’est
cet aspect de l’apoptose que nous allons traiter dans cette brève
revue.
APOPTOSE ET NÉCROSE :
LES DEUX FACES D’UNE MÊME MÉDAILLE ?
L’apoptose et la nécrose sont deux modes de mort cellulaire clas-
siquement décrits au cours de la perte neuronale pathologique
(i.e. ischémie...). Malgré les différences que l’on peut observer
entre ces deux processus, il semble de plus en plus probable
qu’ils empruntent des voies moléculaires communes (2). L’apop-
tose est un processus de mort cellulaire, provoquée dans des
conditions physiologiques. Elle implique, en fin de processus,
une phagocytose par des macrophages ou par d’autres cellules
adjacentes, sans pour autant provoquer une réponse inflamma-
toire. Les changements morphologiques et biochimiques des
cellules apoptotiques se caractérisent par un rétrécissement cel-
lulaire, un bourgeonnement de la membrane, une condensation
de la chromatine, une fragmentation “en échelle” inter- et intra-
nucléosomale de l’ADN, ainsi que par la formation de petites
vésicules nommées corps apoptotiques. Lors d’une mort non
physiologique causée par des substances toxiques ou d’autres
traumatismes, on observe plutôt la nécrose, et cela généralement
dans un groupe de cellules. L’altération des membranes cellu-
laires, l’absence d’utilisation d’ATP, la perte de l’homéostasie
ionique et, enfin, le gonflement et la lyse cellulaires sont des
caractéristiques de la mort nécrotique. Malgré l’ancienneté du
processus, il n’existe pas de détails moléculaires sur le proces-
sus nécrotique, alors que l’apoptose a fait l’objet d’une intense
recherche depuis sa découverte en 1972 (3).
APOPTOSE :VOIES SIGNALÉTIQUES
ET CASCADES DESTRUCTRICES
L’induction du signal apoptotique
Plusieurs causes peuvent initier le processus apoptotique. L’une
des premières voies mises en évidence lors de l’étude du sys-
tème immunitaire est l’activation par différents récepteurs de
surface membranaire de la superfamille des gènes appartenant
au Tumor Necrosis Factor, tels que Fas/APO-1/CD95R ou
TNFR (figure 1). À cette famille de récepteurs correspond une
famille de ligands (fasL,TNF...) qui activent, après fixation sur
leurs récepteurs homologues, un processus apoptotique. Dans
le cas du récepteur Fas, par exemple, l’association du ligand
induit une trimérisation du récepteur permettant ainsi le recru-
tement des protéines cytoplasmiques FADD (Fas Associated
Death Domain), qui se lient aux domaines intracellulaires ou
domaines de mort du récepteur. FADD s’associe ensuite à une
proenzyme appelée procaspase-8, permettant ainsi son oligo-
mérisation et, par le fait même, son autoactivation (4). Les cas-
pases sont les protéases impliquées dans le processus apopto-
tique ; elles sont classiquement divisées en caspases initiatrices,
dont le rôle essentiel est d’activer des procaspases exécutrices
(figure 2), responsables en majeure partie de la dégradation
cellulaire observée au cours de l’apoptose. La caspase-8
correspond à une des quatre caspases initiatrices identifiées à
l’heure actuelle (caspases-8, -9, -10 et -12) (5). D’autres types
de récepteurs, comme les récepteurs à dépendance (p75NTR,
La Lettre du Pharmacologue - Volume 15 - n° 9 - novembre 2001
159
PHARMACOLOGIE
Apoptose et maladies neurodégénératives
!
M. Pelletier*, F. M. Vallette*
*Unité INSERM 419, 44035 Nantes Cedex 01.
RÉSUMÉ.
L’apoptose est un processus de mort cellulaire physiologique dont la dérégulation semble être impliquée dans de nombreuses patho-
logies, notamment dans les maladies neurodégénératives (MND). La transduction du message apoptotique est complexe et requiert plusieurs
molécules à des niveaux divers de la cellule. Dans les MND, l'apoptose semble être l’aboutissement de dérèglements spécifiques à chacune de
ces maladies. Dans le cas de la maladie d’Alzheimer, le peptide β-A1-42 ou les protéines préséniline-1 et -2 sont des acteurs potentiellement
importants de l’apoptose. Dans le syndrome de Parkinson, ce sont plutôt le stress oxydatif et l'exocytotoxicité et, pour l’Huntington,
une mutation sur le premier gène de la protéine huntingtine qui sont impliqués. Dans cette revue, nous traiterons des différents acteurs de
l’apoptose et de leur implication dans ces différentes maladies.
Mots-clés :
Apoptose - Alzheimer - Parkinson - Huntington - Ischémie cérébrale.
L’

DCC, RET…) peuvent induire l’apoptose. Leur mode d’action
n’est pas encore très bien connu, mais il semblerait qu’ils
activent les caspases d’un façon différente des récepteurs de la
famille TNF et de l’apoptosome (6).
L’apoptose peut également être induite par des inhibiteurs de
protéines kinases ou phosphatases, par un stress oxydatif (ROS,
NO), par une irradiation ou d’autres causes pouvant affecter
l’ADN et, ainsi, impliquer la p53, ou encore par un change-
ment dans l’homéostasie calcique, en provenance d’organites
(mitochondries, réticulum endoplasmique [RE]) et/ou de l’es-
pace extracellulaire (après exposition à certains neurotrans-
metteurs). Dans tous ces cas, la mitochondrie joue le rôle d’or-
ganite central dans l’apoptose conduisant à l’activation d’une
autre caspase initiatrice, la caspase-9 (figure 1).De plus, il faut
noter que certaines voies d’induction impliquant des flux cal-
ciques au niveau du RE mettraient en jeu la caspase-12 (7).
La mitochondrie
La mitochondrie a un rôle central dans la transduction du mes-
sage apoptotique : dans la majorité des cas, elle constitue un
passage obligé, mais parfois, seulement un lieu de potentiali-
sation de l’apoptose (figure 1). Au cours de ce type de mort
cellulaire, on observe une perturbation dans la chaîne de trans-
port d’électrons, dans la phosphorylation oxydative et aussi
dans la production d’ATP. Certaines molécules activatrices des
protéases de la famille des caspases, comme le cytochrome c,
les protéines Smac/Diablo et même certaines procaspases (cas-
pases-2, 3 et 9), ou indépendantes des caspases, l’AIF (Apop-
tosis-Inducing Factor), sont libérées de la mitochondrie après
l’induction d’apoptose et amplifient la transduction du signal
de mort. La quantité de cytochrome c contenue dans la cellule
pourrait déterminer s’il y a apoptose ou nécrose ; avec assez de
cytochrome c pour maintenir le transport d’électrons, la
consommation d’oxygène et la production d’ATP à un niveau
assez élevé, la cascade apoptotique peut avoir lieu. En revanche,
s’il existe une chute trop importante de la production énergé-
tique, un processus de type nécrotique sera favorisé (2).
Lors de l’apoptose, on remarque souvent un effondrement du
potentiel de membrane mitochondrial (∆Ψm), où serait impli-
qué le pore mitochondrial de transition de perméabilité (PTP).
Il se composerait d’un ensemble de protéines réparties dans les
deux membranes de la mitochondrie. On retrouve dans la mem-
brane externe le VDAC (Voltage-Dependent Anion Channel)
ainsi que le PBR (Peripheral Benzodiazepin Receptor) et, dans
160
La Lettre du Pharmacologue - Volume 15 - n° 9 - novembre 2001
PHARMACOLOGIE
Stimulus (FasL)
Récepteur de mort
(Fas)
Stimulus apoptotiques
intrinsèques
Apoptosome
Stress extracellulaires
(privation de sérum, flux calcique
infection virale, irradiation, etc.)
APOPTOSE
ATP
Clivage de DFF-45, lamine Calpaïnes
Procaspase-9
Caspases exécutrices
(caspase-3)
Bid Bci-2
Ca2+
Bax
p53 ?
?
?
caspase-9 activée
(caspase initiatrice)
caspase-8
(caspase
initiatrice)
FADD
Figure 1. Représentation schématique des différentes voies de signalisation
apoptotiques.
Différents types de stress extracellulaire peuvent provoquer des stimulus apopto-
tiques intrinsèques. Ces derniers transitent par des effecteurs comme Bax, via p53,
qui induisent l’éjection du cytochrome c de la mitochondrie. Il y a ensuite formation
d’un apoptosome, puis l’activation des caspases exécutrices et la destruction cellu-
laire. Il existe en plus des voies alternes, comme celles passant par Bid ou encore
par le calcium libre et les calpaïnes. L’activation des différents récepteurs provoque
une autoactivation des caspases-8, qui activent à leur tour des caspases exécutrices.
Figure 2. Représentation schématique des trois grands
groupes de caspases.
Les caspases initiatrices les plus connues sont les caspases-
8 et -9. Elles sont spécifiques par la séquence de leur
substrat. Les caspases inflammatoires sont moins impor-
tantes dans l’apoptose que dans les réactions inflamma-
toires. Le troisième groupe, les caspases exécutrices, est
responsable de l’amplification de la cascade apoptotique
et de la destruction de la cellule.
Caspases
initiatrices
Caspase-8
Caspase-9
Caspase-10
Caspase-12
IETD
LEHD
?
?
Caspases
inflammatoires
Caspase-1
Caspase-4
Caspase-5
Caspase-13
YVAD
(W/L) EHD
(W/L) EHD
?
Caspases
exécutrices
Caspase-2
Caspase-3
Caspase-6
Caspase-7
DEVD
DEVD
VEID
DEVD
?
?
Caspases Substrats Caspases Substrats
Caspases Substrats

la membrane interne, l’ANT (Adenine Nucleotide Translo-
cator) (8). Cependant, seules des molécules ≤1,5 kD peuvent
passer par ce canal. Le cytochrome c (14 kD) et l’AIF (57 kD)
ne pouvant pas transiter par ce canal, deux hypothèses peuvent
être émises concernant leur éjection de la mitochondrie. La pre-
mière considère que le cytochrome c passerait par un canal
ouvert, la membrane externe restant donc intacte. La deuxième
hypothèse impliquerait le gonflement de la matrice et donc, la
rupture transitoire de la membrane externe (3).
Les oncoprotéines de la famille Bcl-2
Il existe une famille de gènes codant pour des protéines impli-
quées dans la régulation de l’apoptose, de par la formation
d’homo- et d’hétérodimères, qui agiraient sur la mitochondrie.
Ces protéines constituent un premier moyen de contrôle de
l’apoptose. Leur famille, d’une vingtaine de membres, se divise
en deux grandes classes : les protéines antiapoptotiques comme
Bcl-2, Bcl-XL,Mcl-1, CED-9, etc., et celles proapoptotiques,
telles que Bax, Bak, Bid, Bad, Bcl-XS,etc. (figure 3). C’est
l’équilibre entre les protéines antiapoptotiques et les protéines
proapoptotiques qui pourrait déterminer le sort de la cellule.
Toutes les protéines de cette famille possèdent des motifs
consensus qui sont, en fait, des domaines d’homologie de Bcl-
2, BH1 à BH4 (Bcl-2 homology domain). Une très grande
majorité des protéines favorisant la survie cellulaire compor-
tent les domaines BH1 et BH2. En revanche, parmi les pro-
téines incitant à la mort cellulaire, on trouve deux types de pro-
téines. La sous-classe Bax comprend des éléments comme Bax,
Bak et Bok, qui comportent les domaines BH1, BH2 et BH3.
Une dernière sous-classe est composée de protéines comme
Bik, Bad, Bid, qui ne contiennent que le domaine BH3 (9).
Bcl-2 est exclusivement une protéine membranaire ; elle se fixe
sur la face cytoplasmique de la membrane externe mitochon-
driale (en majorité), du réticulum endoplasmique (RE) et du
noyau, grâce à son domaine d’ancrage situé à l’extrémité
C-terminale, caractéristique de la majorité des protéines anti-
apoptotiques. Cette protéine peut inhiber la libération du
cytochrome c (10) et contrôler également les flux calciques.
Bcl-XLse répartit différemment dans la cellule : une portion
significative est présente dans le cytosol, mais on en retrouve
également sur les membranes. Sa structure ressemble au
domaine d’insertion membranaire de toxines bactériennes, en
particulier les hélices α5 et α6, situées à cheval sur les domaines
BH1 et BH2. Les protéines possédant ces deux domaines
seraient susceptibles de former des pores dans les membranes.
Bax est une protéine cytosolique, mais elle devient membra-
naire suite à un signal apoptotique. Contrairement à Bcl-2,
l’extrémité C-terminale de Bax ne semble pas être un signal
d’adressage ou d’ancrage (11). Dans le cas de Bid, il s’agit
d’une protéine cytosolique lorsqu’elle est non clivée, en condi-
tions non apoptotiques. Après une activation de type Fas/FasL,
Bid est clivée directement par la caspase-8. Cette forme tron-
quée (tBid) se translocalise et semble se fixer solidement sur
la mitochondrie.
L’apoptosome
La présence du cytochrome c dans le cytosol revêt une grande
importance dans la suite des événements conduisant à la mort
cellulaire programmée (figure 1). Il participe à l’élaboration de
l’apoptosome (l’association transitoire de molécules) en un agré-
gat, résultant en l’activation de protéases exécutrices de l’apop-
tose. À nouveau, plusieurs hypothèses sur sa composition ont
été émises. D’après l’équipe de Dixit, sous forme inactive, il se
composerait d’une protéine antiapoptotique de la famille
Bcl-2, Bcl-XL,liée à son extrémité C-terminale à la mitochon-
drie et, en N-terminale, à une molécule activatrice, l’Apaf-1. La
procaspase-9 se lierait sur l’extrémité N-terminale d’Apaf-1 pour
former un complexe ternaire (12). Lors de l’activation de molé-
cules proapoptotiques, comme Bax ou Bak, le détachement de
Bcl-XLde l’apoptosome semble favoriser la libération du cyto-
chrome c de l’espace intermembranaire de la mitochondrie vers
le cytosol. S’ensuit la mobilisation du cytochrome c sur l’apop-
tosome. Ainsi, l’apoptosome actif est constitué d’un regroupe-
ment d’au moins quatre molécules : l’Apaf-1, l’ATP ou le dATP,
le cytochrome c et la procaspase-9.
Une deuxième hypothèse avance que l’Apaf-1 hydrolyserait
tout d’abord l’ATP et favoriserait son oligomérisation avec le
cytochrome c en un complexe multimérique. La caspase-9 serait
ensuite recrutée sur cet amas dans un ratio de un pour un avec
La Lettre du Pharmacologue - Volume 15 - n° 9 - novembre 2001
161
PHARMACOLOGIE
Figure 3. Protéines de la famille Bcl-2.
Les protéines Bcl-2, Bcl-XLet Mcl-1 sont antiapoptotiques. Elles
comportent les domaines d’homologie BH1 à BH3 dans la majorité
des cas, ainsi qu’une extrémité C-terminale transmembranaire (TM).
Les protéines proapoptotiques se divisent en deux sous-classes : Bax,
Bak et Bad possèdent les domaines d’homologie BH1 à BH3. En
revanche, Bid, Bim et Bik font partie de la sous-classe BH3 et ne com-
portent que ce domaine d’homologie.
α1α2α3α4α5α6α7
a
a : docking ; b : phosphorylation ; c : ligand ; d : pore.
bc d
BH4 ***BH3 BH1 BH2
Bid
Bik
Bim
Bad
Bax
Bax
Bak
Bcl-2
Bcl-XL
Bcl-Xs

l’Apaf-1 (13). Une fois recrutée sur l’Apaf-1, la caspase-9 est
activée et transduit la cascade apoptotique. Contrairement aux
caspases exécutrices, elle n’aurait aucun besoin de se cliver en
plusieurs parties pour être effective.
Les caspases exécutrices
L’activation des caspases initiatrices (caspases-8 et -9) permet
l’initiation de la cascade de destruction cellulaire. Les acteurs
principaux de cette phase sont les caspases exécutrices. Elles
sont formées de trois sous-unités, mais sont présentes dans le
cytosol des cellules non apoptotiques sous forme de zymogènes
(figure 4). La plus grande sous-unité contient le site actif de la
caspase, QACXG, et la petite sert à la réorganisation de la cas-
pase en une enzyme active. Il existe également un prodomaine,
souvent plus court chez les caspases exécutrices que chez les
initiatrices, éliminé lors de l’activation. Lorsqu’il y a induction
d’apoptose, la grande et la petite sous-unité se réorganisent en
hétérotétramères. Dans le cas de la caspase-3, il a été
montré que ces deux clivages étaient successifs. Le premier se
ferait à un site spécifique pour les caspases-8, IETD, soit entre
la petite et la grande sous-unité. L’élimination du prodomaine
viendrait en deuxième lieu, au site ESMD, spécifique pour les
caspases-3, ce qui indiquerait un autoclivage (14).
Quatorze caspases ont été identifiées à l’heure actuelle. On les
divise en trois groupes : quelques-unes d’entre elles sont impli-
quées dans les réactions inflammatoires (1, 4, 5, 13), les autres,
dans la transduction de l’apoptose (figure 2). Les caspases-8,
-9, -10 et -12 (initiatrices) se situent en amont des caspases-2,
-3, -6 et -7 (exécutrices), mais souvent, on les classe par spé-
cificité pour leurs substrats (tableau I
[seules les caspases les plus
connues sont représentées dans ce tableau]
)(4).
Il existe, au niveau de cette phase d’exécution, un autre moyen
de contrôle de l’apoptose que celui de la famille de protéines
Bcl-2. En fait, il s’agit d’une autre famille de protéines inhibi-
trices de caspases, les IAP (Inhibitor of Apoptosis Protein) (15).
Elles possèdent un domaine répété et hautement conservé, d’en-
viron 70 acides aminés : BIR (Baculoviral Inhibitory Repeat).
Six membres des IAP humaines ont été identifiés, soit NAIP,
c-IAP1/HIAP-2, c-IAP2/HIAP-1, XIAP/hILP, Survivin et
BRUCE. Elles n’agissent que sur les caspases-3 et -9. La liai-
son et l’inhibition des caspases-3 et -7 par XIAP, c-IAP1,
c-IAP2 et Survivin nécessitent leur transformation en une forme
active. Dans le cas de la caspase-9, puisqu’elle ne nécessite pas
de clivage pour être active (16),les IAP fixent aussi bien la pro-
forme que celle activée.
Les caspases exécutrices possèdent plusieurs substrats, comme
d’autres caspases, mais également des composants cellulaires
tels que la lamine, certaines DNAses, SREBP, PARP, etc.
Les calpaïnes dans l’apoptose
Les voies classiques de l’apoptose impliquent pratiquement
toujours les caspases. Cependant, il existe des voies compre-
nant également des calpaïnes. Ce sont des protéases de la
famille des cystéinyl/thiol activées par le calcium, qui com-
portent six isoformes tissus-spécifiques (n-calpaïnes) et deux
isoformes ubiquistes (µ-calpaïne et m-calpaïne). La µ-calpaïne
(ou calpaïne I, ou encore CANP-I) possède une affinité rela-
tivement élevée pour le calcium et se situe d’une façon pré-
dominante dans les neurones, plus précisément dans les den-
drites et les corps cellulaires. La m-calpaïne (ou calpaïne II,
ou encore CANP-II) lie le calcium avec une affinité plutôt
faible et se situe, pour sa part, dans les axones et les cellules
gliales.
La proenzyme de la calpaïne est constituée en hétérodimères :
une sous-unité catalytique de 80 kD, spécifique à chaque iso-
mère, et une sous-unité régulatrice de 30 kD commune à tous
les isomères. Le site catalytique contient des résidus cystéine
et histidine. Leur activation se fait à la suite d’une augmenta-
tion de calcium libre intracellulaire et leur inhibition par une
protéine inhibitrice (la calpastatine) ou par une diminution du
niveau de calcium. Une fois activées, la sous-unité de 80 kD
est clivée en 78 kD puis 76 kD, et celle de 30 kD, en 18 kD.
Malgré le fait que leur mode d’activation soit en partie connu
et qu’elles intègrent la famille des protéases à cystéine active,
tout comme les caspases, il n’est pas évident de savoir si ces
dernières travaillent de concert ou indépendamment au cours
de l’apoptose.
162
La Lettre du Pharmacologue - Volume 15 - n° 9 - novembre 2001
PHARMACOLOGIE
Figure 4. Mode d’activation de la procaspase-3.
Le zymogène de 32 kD est clivé entre la petite (p12) et la grande (p17)
sous-unité, au site IETD. La caspase-3 s’autoactive ensuite en se cli-
vant au site ESMD, pour éliminer le prodomaine de la grande sous-
unité. Sa forme active finale est un hétérotétramère contenant deux
sites actifs QACXG dans chacune des p17 et deux p12 régulatrices.
p12
Site actif
QACXG
ESMD28 IETD175 S
Procaspase-3
32 kD
p3 p17 p12 C-terminal
(277 a.a.)
N-terminal
p3 +p17
p17
p12
p20
Pro-
domaine Grande
sous-
unité
Petite
sous-
unité
p12
p17

APOPTOSE ET MALADIES NEURODÉGÉNÉRATIVES
Maladie d’Alzheimer
La maladie d’Alzheimer (MA) touche 50 à 60 % des personnes
atteintes de maladies neurodégénératives. Il existe des formes
héréditaires (génétiques) et sporadiques de cette maladie. Elle
se caractérise par une perte progressive de la mémoire à court
terme puis à long terme, par une diminution des capacités intel-
lectuelles et, parfois, par des troubles moteurs. Les régions du
cerveau les plus touchées sont le cortex rhinal et l’hippocampe,
impliqués dans les processus de mémorisation, au sein desquels
les neurones cholinergiques sont particulièrement sensibles à
l’apoptose. Les caractéristiques majeures retrouvées dans ces
zones de mort sont un dépôt important de peptides
β-amyloïde (βA), composant des plaques séniles, ainsi qu’une
dégénérescence neurofibrillaire. Dans les formes héréditaires
de la MA, on retrouve une mutation de cette protéine sur le
chromosome 21 (mutation de type Swedish) (17).
Plusieurs protéines ont été identifiées comme faisant partie du
mécanisme de mort, mais on ne connaît pas encore complète-
ment leur rôle. Un des composants principaux des plaques
séniles, le βA, résulte du clivage de l’APP (Amyloid Precursor
Protein). La majorité de l’APP est clivée par l’α-sécrétase, ce
qui génère un grand fragment se libérant de la membrane.
L’APP peut également être clivée sur deux autres sites, βet γ,
à l’intérieur d’un domaine intramembranaire. Le βA1-40 ainsi
formé et sécrété dans l’espace extracellulaire est présent dans
le cerveau dans des conditions normales. En revanche, le βA1-42
tend à polymériser en fibrilles et se retrouve en grande quan-
tité dans la MA. Au cours de l’apoptose, l’APP peut être clivée
par la caspase-3 à trois endroits possibles : deux sur le domaine
extracellulaire et un (VEVD) dans la queue intracellulaire.
Après un clivage au site VEVD, l’APP obtenue semble plus
sensible à la production de βA. De plus, lors de l’induction de
l’apoptose, par ajout de peptides βA1-42 au sein de cultures neu-
ronales primaires, on constate une diminution de la protéine
antiapoptotique Bcl-2, ainsi qu’une augmentation de la
protéine proapoptotique Bax (18).
Le βA est également impliqué dans d’autres phénomènes
conduisant à la mort neuronale, comme une augmentation
substantielle de calcium dans le cytosol. La caspase-3 et la
calpaïne étant toutes deux activées par le calcium, et l’APP
étant elle-même un substrat de ces protéases, on peut en déduire
que le calcium est éventuellement un amplificateur de la voie
apoptotique.
Les présénilines 1 et 2 sont également impliquées dans les
formes sporadiques et génétiques de cette maladie. Dans cette
dernière forme, on retrouve une mutation de la préséniline-1
(PS1) sur le chromosome 14 et de la préséniline-2 (PS2) sur le
chromosome 1. Ordinairement, les protéines PS1 et PS2, de 52-
54 kD, sont localisées dans les membranes du noyau, du RE,
de l’appareil de Golgi et très peu dans les membranes plas-
miques des neurones, puis elles sont clivées en protéines
matures de 25-28 kD et 16-19 kD. Cependant, au cours de
l’apoptose, la caspase-3 reconnaît d’autres sites qui lui sont spé-
cifiques et donne lieu, pour la PS2, à des protéines de 34 kD en
N-terminale et de 20 kD en C-terminale. Ces nouveaux pep-
tides sont retrouvés en très grand nombre dans les formes fami-
liales de la MA (FMA). De plus, on constate que la quantité de
peptide βA1-42 est augmentée dans la FMA, grâce aux PS
mutées (19).
La Lettre du Pharmacologue - Volume 15 - n° 9 - novembre 2001
163
PHARMACOLOGIE
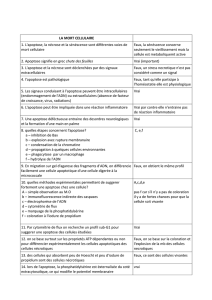
Tableau I. Spécificité des sites de clivage interne des caspases les plus connues ainsi que ceux correspondant à leurs substrats.
Caspases Autres noms X du site actif Site de clivage Motifs de clivage Substrats
(QACXG) entre la grande de différents correspondants
et la petite sous-unité substrats
Caspase-1 ICE R WFKD-S ; FEDD-A YVHD-A Pro-IL-1ß
Caspase-2 Nedd2, ICH-1 R DQQD-G ; EESD-A
Caspase-3 CPP32, Yama, R IETD-S DEVD-(G/N) ; PARP/DNA-PK
apopaïne DGPD-G ; U1-70 kD
DMQD-N ; Protéine kinase C
DELD-S ; D4-GDP DI
DEPD-S ; SREBP-1 et 2
DEAD-G Rb
DXXD Huntingtine
Caspase-4 ICErel II, TX, ICH-2 R WRVD-S ; LEED-A
Caspase-5 ICErel III, TY R WRVD-S ; LEAD-S
Caspase-6 Mch2 R DVVD-N ; TEVD-A VEID-N Lamine A
Caspase-7 Mch3, ICE-LAP3, R IQAD-S DEVD-G ; PARP
CMH-1 DEPD-S SREBP-1 et 2
Caspase-8 MACH, FLICE, Q VETD-S ; LEMD-L (I/L) ETD- ?
Mch5
Caspase-9 ICE-LAP6, Mch6 G DQLD-A LEHD- ?
Caspase-10 Mch4 Q SQTD-V ; IEAD-A
 6
7
8
6
7
8
1
/
8
100%