Aucun titre de diapositive

PROCESSUS
DE CANCERISATION
AU NIVEAU CELLULAIRE
F. PEDEUTOUR
DCEM1-3/10/2011

CANCER : MALADIE DE LA CELLULE
Cause : dérégulation du programme
génétique cellulaire
PATHOLOGIE DE L’ADN
Conséquence : prolifération incontrôlée de
cellules anormales avec envahissement
local ou à distance

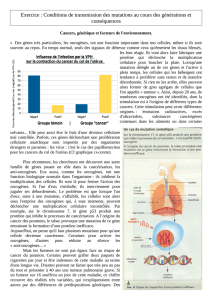
I. Rappel : prolifération, différenciation,
vieillissement et mort des cellules normales
Cycle cellulaire normal: bien défini et contrôlé
Etapes-clés: G1, S, G2, M
Dans certaines circonstances bien définies (programmées
ou en réponse à un signal activateur), la cellule se divise
pour donner une autre cellule identique lors de la
mitose (M)
Etapes de contrôle :
arrêts temporaires pour réparation de l’ADN si lésion,
ou
arrêts définitifs si trop d’anomalies non réparables

I. Prolifération, différenciation, vieillissement
et mort des cellules normales
Entrée dans le
cycle cellulaire :
-signalisation
en cascade
-action des
cyclines
Cycle cellulaire
avec étapes de
contrôle
Systèmes de
réparation
ou d’arrêt du
cycle
G 0
cellule quiescente
tat de repos
G 1
tapes de prparation
quelques heures quelques jours
Ssynthse : 7 heures
duplication ADN
G 2
M
3 heures
mitose
1 heure
P53 rgulateur+++
G 0G 0
cellule quiescente
tat de repos
G 1G 1
tapes de prparation
quelques heures quelques jours
SS synthse : 7 heures
duplication ADN
G 2G 2
MM
3 heures
mitose
1 heure
P53 rgulateur+++
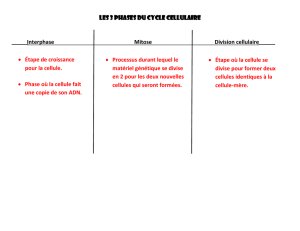
réplication
Préparation à la mitose
Division cellulaire :
2 cellules filles

I. Prolifération, différenciation, vieillissement
et mort des cellules normales
Si nécessaire, la cellule normale met en œuvre :
des outils de prévention
en vue de la limitation de la survenue
d’anomalies transmissibles aux cellules filles :
-Systèmes de réparation de l’ADN
ou
- « suicide » = « apoptose » (mort cellulaire
programmée)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
1
/
40
100%