hematologie - Ecole Rockefeller

HEMATOLOGIE
Pierre FAURIE

Généralités
•Hématologie: étude des maladies du sang, et,
par extension, des maladies de la moelle
osseuse et des organes hématopoïétiques
secondaires (rate, ganglions)
•Cellules sanguines: globules blancs
(polynucléaires, lymphocytes, monocytes),
globules rouges, plaquettes.
•Production au sein de la moelle osseuse, grâce
aux propriétés des cellules souches
hématopoïétiques.

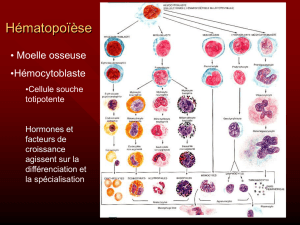
Cellules sanguines
Lignée myéloïde Lignée lymphoïde
Plaquettes
Globules rouges
Monocytes
Polynucléaires
neutrophiles
basophiles
éosinophiles
Lymphocytes B
Plasmocytes
Lymphocytes T
lymphocytes T CD4+
lymphocytes T CD8+
Lymphocytes NK

•cellule souche
totipotente
•cellules
souches
pluripotentes
•cellules
souches
orientées
•cellules
matures
Hématies Plaquettes Neutrophiles Monocytes Eosinophiles Basophiles Lymphocytes
SANG
Moelle osseuse

Exploration de la moelle
osseuse
•Myélogramme:
–sous anesthésie locale (AL)
–au niveau sternal ou iliaque
–habituellement: peu douloureux et sans complication
•Biopsie ostéomédullaire:
–sous AL+ MEOPA (idéalement)
–uniquement au niveau iliaque
–risque hémorragique (faible)
–plus douloureux et plus long que le myélogramme:
réservé aux échecs de ce dernier, ou d’emblée dans
certaines indications (bilan d’extension des
lymphomes en particulier)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
1
/
42
100%