M1ParisV

IN VIVO STUDIES IN ANIMALS :
PRECLINICAL PHARMACOLOGY
An essential step between molecular and human studies for
understanding the pathophysiological mechanisms involved in human
diseases and the mechanisms of action of pharmacological agents.

DEVELOPMENT OF EXPERIMENTAL MODELS
OF CARDIAC AND RENAL DISEASES
•Post-Ischemic Heart Failure (Coronary ligation)
(Pons et al., Clin. Exp. Physiol. Pharmacol., 2003; J. Cardiovasc.
Pharmacol., 2003; Richer et al., Circulation, Abstract, 2003)
•Myocardial Ischemia-Reperfusion Injury (reversible coronary oclusion)
(Richer et al., FASEB J, 2005)
•Renovascular Hypertension (clip on renal artery)
•Experimental Aldosterone-induced Cardiac Remodeling
•Insulinoprive Diabetes (Streptozotocin)
(Huang et al., PNAS, 2003)

ANIMAUX TRANSGENIQUES ET
MALADIES CARDIOVASCULAIRES
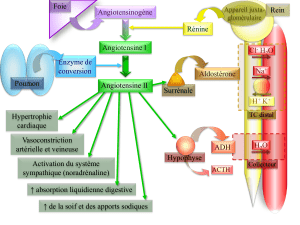
Ce sont des modèles avec une ou plusieurs modifications
ciblées d’une hormone ou d’un second messager impliqués
dans la régulation cardiovasculaire

GENETICALLY MODIFIED ANIMALS
Generation of genetically altered animals :
- Models of gene inactivation : Tissue Kallikrein,ApoE.
- Models of gene inactivation and duplication, genetic titration :
ACE, Angiotensinogen,Tissue Kallikrein.
Candidate genes : Vasoactive peptide systems, Ionic transporters.
Breeding of mutated strains :
- Importance of genetic backgrounds
(Trabold et al., Hypertension, 2OO2)
- Need of individual genotyping
- Use of control littermates
- Role of environmental factors : housing, diet.

Survie des rats Brown Norway (BN) et
Fischer344 (F) après traitement au L-NAME.
Souche Fischer 344 susceptible
Développement d’AVC
Effets variables du traitement au L-NAME
selon les souches de rats
Souche Brown Norway résistante
Absence d’AVC
BN
F
010 20 30 40 50 60 70 80 90
Time (days)
0
.2
.4
.6
.8
1
Survival rate (%)
Pression artérielle des deux souches
après traitement au L-NAME.
0
50
100
150
200
250
300
pression
artérielle
(mmHg)
PA 15j
153
187
132
163
PA 30j
157
278
134
210
FT
FL
BNT
BNL
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
1
/
45
100%