catalyseL3

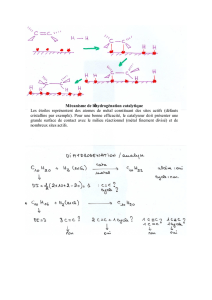
COURS DE CATALYSE
INTRODUCTION (1h30)
Catalyse hétérogène
(7h30) M.-C. GAZEAU
Catalyse homogène acide-base et enzymatique
(6h) F. RAULIN
Catalyse homogène par les métaux de transitions
(7h30) S. CONDON

LA CATALYSE - INTRODUCTION
Les réactions catalytiques jouent un rôle important dans notre vie :
dans notre corps, dans notre quotidien
Réactions biologiques :
respiration,
transport et assimilation des aliments,
photosynthèse des plantes,
…
Processus industriels majeurs (fabrication des produits chimiques usuels):
engrais (synthèse de l’ammoniac NH3par processus catalytique)
colles, plastiques, peintures (synthèse du méthanol CH3OH par processus catalytique)
raffinage du pétrole brut (forme non utilisable) : obtention de produits utilisables
carburants (Gaz de Pétrole Liquéfié, essences, gasoils),
combustibles (fioul),
produits spéciaux (cires ou paraffines),
…
Voitures, usines engendrent des composés nocifs :
élimination ou transformation de ces derniers en produits inoffensifs

I) ETUDE D’UNE TRANSFORMATION CHIMIQUE - RAPPELS
I-1) Etude thermodynamique :
permet de répondre aux questions :
est ce que la réaction est possible ou non ?
est ce que le système peut atteindre une position d’équilibre stable ?
apprend :
qu’une fonction locale ou fonction d’état est associée à une quantité de matière
(énergie)
qu’une réaction chimique se traduit par une perte ou un gain d’énergie
ne traite que les différences d’énergie entre l’état initial et l’état final
ainsi la variation d’énergie lors de la transformation chimique ne dépend pas du trajet

I-1) Etude thermodynamique (suite) :
Grenoble Gène
Altitude = fonction d’état locale définie pour les 2 villes
214 m de différence d’altitude
Grenoble Gène
descente
Montée !!!
Mais ne donne pas de renseignement sur la route ….

I-2) Etude cinétique :
Permet de répondre aux questions (lorsqu’une réaction a été déterminée
comme possible par la thermodynamique) :
à quelle vitesse la réaction va-t-elle avoir lieu ?
est ce que l’équilibre est rapidement atteint ou non ?
La vitesse de la réaction dépend de la barrière énergétique (ou énergie
d’activation) que les réactifs doivent surmonter lors de leur
transformation en produits.
La hauteur de cette barrière est déterminée par le mécanisme de la
réaction.
passer par les Alpes
passer par Marseille puis longer la mer
Des routes plus faciles que d’autres :
Grenoble Gène
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
1
/
85
100%