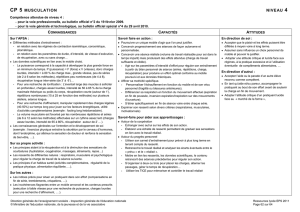
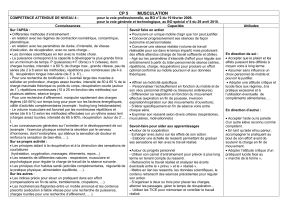
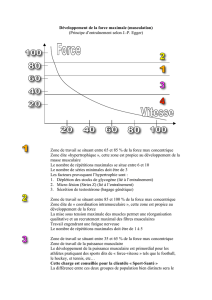
dea.pps - CRIStAL

1
Recherche de répétitions distantes dans
les séquences
Etudiant : Laurent NOE Encadrant : Gregory KUCHEROV

2
Plan
1. Introduction au problème
2. Les programmes existants
3. La méthode adoptée
4. L’algorithme
5. Résultats obtenus et extensions envisagées
6. Conclusion

3
1. Introduction
L’ADN
•La molécule
• L’information contenue
Extraction de l’information (séquençage)
Gènes et fonctions
Aspects automatisables

4
Recherche de répétitions
• Problème connu de l’algorithmique du texte
• Spécificité de l’ADN : répétitions approchées
•Sous-répétitions exactes (graines)
•Approche choisie

5
Evolution des occurrences d’une
répétition
s
1 2 3 123
d
1
3
4
5
1435
i
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
1
/
28
100%