DS n°5 - CPGE Brizeux

PC A - PC B CHIMIE - DS n°5 - Correction
1
Partie 1 :
Le vert malachite
D’après le concours ENSTIM 2009, filière PCSI
Q1. La loi de Beer-Lambert énonce que l’absorbance (sans unité) d’une solution contenant une
espèce est proportionnelle à la concentration de cette espèce, c, en mol.L
-1
, et à la largeur de cuve
traversée l en m, le coefficient de proportionnalité étant ε, le coefficient d’absorption (ou d’extinction)
molaire en mol
-1
.L.m
-1
: A = ε
εε
ε l c
Si plusieurs espèces absorbent le rayonnement, leur absorbances s’additionnent.
Q2. La courbe représentative de l’absorbance A en fonction de la longueur d’onde
λ
est le spectre
d’absorption. On choisit en général la longueur d’onde pour laquelle l’absorbance est maximale,
c’est-à-dire ε(λ) maximal, lorsque l’on cherche à vérifier la loi de Beer-Lambert, pour mesurer des
variations importantes d’absorbance en fonction de la concentration et avoir ainsi une meilleure
précision.
Q3. A l’aide, par exemple, d’une pipette jaugée deux traits, que l’on peut préalablement rincer avec
la solution mère, on prélève 10,0 mL de la solution mère que l’on place dans une fiole jaugée de 100
mL, que l’on complète jusqu’au trait de jauge avec de l’eau distillée ; on bouche puis on homogénéise.
On obtient ainsi la solution fille, diluée d’un facteur 10 par rapport à la solution mère.
Q4. Les points sont alignés donc la loi de Beer-Lambert est vérifiée. (Remarque : il faudrait la
valeur du coefficient de corrélation pour justifier complètement cette affirmation.) La valeur du
coefficient d’absorption molaire ε du vert malachite est égale à celle du coefficient directeur.
A.N. ε = 58.10
3
mol
-1
.L.m
-1
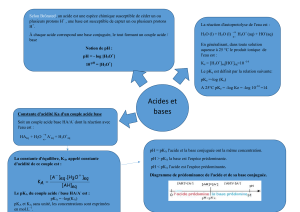
Q5. Schéma détaillé et légendé des titrages pH-métrique et conductimétrique. Cf cours. Ne pas
oublier de dessiner le conductimètre et le pHmètre et surtout de nommer les électrodes ! (pHmétrie :
électrode de mesure = électrode de verre + électrode référence = électrode au calomel saturé par
exemple ; conductimétrie : une seule électrode plonge dans le bécher : la cellule conductimétrique.)
Q6. Compte-tenu des constantes d’acidité, avant la première équivalence, H
3
O
+
et HSO
4-
de pKa 0
et 2 sont dosés non successivement car l’écart entre leur pKa est inférieur à 4, selon :
H
3
O
+
+ HO
-
= 2 H
2
O K° = Ke
-1
= 10
14
et HSO
4-
+ HO
-
= SO
42-
+ H
2
O K° =
Ke
Ka
= 10
12
Puis entre les deux équivalences, NH
4+
, de pKa 9,2, est dosé seul, successivement des deux autres car
∆pKa > 4 : NH
4+
+ HO
-
= NH
3
+ H
2
O K° =
Ke
Ka
= 10
4,8
.
Q7. On lit graphiquement les volumes des points d’équivalence : v
e1
= 10,0 mL et v
e2
= 25,0 mL.
Les deux courbes confirment ces valeurs mais la première équivalence est plus visible sur la courbe de
dosage pHmétrique, le premier saut est plus grand, et la deuxième est plus visible sur la courbe de
dosage conductimétrique : changement de pente plus net.
Q8. Effectuons les bilans de matière aux deux équivalences :
Samedi 13 février 2010
DS n°5
Chimie Organique et Acido-Basicité
Durée : 4 heures
CORRECTION

PC A - PC B CHIMIE - DS n°5 - Correction
2
C
B
v
e1
= 2 C
A
V
A
donc C
A
=
V 2 vC
A
e1B A.N. C
A
= 5,0.10
-2
mol.L
-1
.
C
B
(v
e2
- v
e1
) = C’
A
V
A
donc C’
A
=
V ) v- (vC
A
e1e2B A.N. C
A
= 1,5.10
-1
mol.L
-1
.
Q9. Après la deuxième équivalence, la conductivité augmente car il y a ajout d’ions Na
+
et HO
-
, en
excès, qui ne réagissent donc plus.
Q10. Il aurait été possible d’utiliser un indicateur coloré pour repérer le 1
er
point d’équivalence car le
saut de pH est ample, l’hélianthine ou le rouge de méthyle virerait bien à la goutte près à la première
équivalence car le saut comprend sa zone de virage. Pour le 2
ème
point d’équivalence aucun indicateur
ne conviendrait car le saut est trop petit et n’englobe aucune zone de virage. Le jaune d’alizarine ou
(pire) la phénolphtaléine vireraient sur une fourchette de volume trop grande.
Partie 2 :
pH du sang et effort musculaire
D’après CCP TSI 2005
Le pH du sang est tamponné par le couple H
2
CO
3
/HCO
3-
. Dans le sang d’une personne au repos, les
concentrations en HCO
3-
et H
2
CO
3
sont respectivement de 0,027 mol.L
-1
et 0,0014 mol.L
-1
.
1) Le terme tamponné signifie que le pH varie peu par ajout modéré d’acide, de base, ou par dilution.
Les espèces carbonées présentes dans le sang proviennent de la dissolution en solution aqueuse du
dioxyde de carbone.
2) Le sang contient HCO
3-
et H
2
CO
3
(également écrit H
2
O,CO
2
) :
H
3
O+
pK
A
H
2
O
CO
32-
0
10,3
HCO
3-
HO
-
14
H
2
O
HCO
3-(aq)
+ H
2
CO
3(aq)
= H
2
CO
3(aq)
+ HCO
3-(aq)
K°
1
= 1
Dans le milieu sont présents 3 acides et 2 bases, 6 réactions sont
donc envisageables.
(1)
HCO
3-
6,4
H
2
O, CO
2
La réaction (1) est la réaction prépondérante, c'est l'équilibre de contrôle.
Cette réaction ne modifie pas les concentrations des espèces en présence.
Les concentrations à l'équilibre sont donc égales aux concentrations initiales.
HCO
3-(aq)
+ HCO
3-(aq)
= H
2
CO
3(aq)
+ CO
32-(aq)
K°
2
= K
A2
/K
A1
= 10
-3,9
.
H
2
O
(l)
+ H
2
O
(l)
= HO
-(aq)
+ H
3
O
+(aq)
K°
6
= K
e
= 10
-14
.
(2)
(4)
HCO
3-(aq)
+ H
2
O
(l)
= CO
32-(aq)
+ H
3
O
+(aq)
K°
5
= 10
-10,3
.
HCO
3-(aq)
+ H
2
O
(l)
= H
2
CO
3(aq)
+ HO
-(aq)
K°
4
= 10
-7,6
.
(5)
(6)
(3) H
2
CO
3(aq)
+ H
2
O
(l)
= HCO
3-(aq)
+ H
3
O
+(aq)
K°
3
= 10
-6,4
.
On peut alors obtenir le pH : pH = pK
a1
+ log[HCO
3-
]/[H
2
CO
3
] = 7,7.
Par ailleurs, on peut négliger les réactions de HCO
3-
et H
2
CO
3
sur l’eau.

PC A - PC B CHIMIE - DS n°5 - Correction
3
3) Pour montrer que l’espèce CO
32-
est négligeable, on va étudier la réaction (2) :
2 2
2
2
2
(0,0014 )
(0,027 2 )
x x
Kx
+
° = −
soit x
2
= 6,4.10
-5
mol.L
-1
. Cette concentration est bien négligeable
devant les autres concentrations en espèces carbonées.
4) Au cours d’efforts physiques importants, il se forme de l’acide lactique, noté AH dans les muscles.
Cet acide passe dans le sang. Réaction qui peut avoir lieu entre les espèces contenues dans le sang et
l’acide lactique : CH
3
-CHOH-COOH
(aq)
+ HCO
3-(aq)
= CH
3
-CHOH-COO
-(aq)
+ H
2
CO
3(aq)
[
]
[ ]
3 2 3
1
3 3
a
a
CH CHOHCOO H CO
K
K
K
CH CHOHCOOH HCO
−
−
°= =
= 321
5) Après un effort violent, l’acide lactique passe dans le sang à raison de 0,003 mol.L
-1
.
H
3
O+
pK
A
H
2
O
CO
32-
0
10,3
HCO
3-
HO
-
14
H
2
O
HCO
3-
6,4
H
2
O, CO
2
A
-
3,9
AH
La réaction prépondérante est une réaction quasi-totale :
CH
3
-CHOH-COOH
(aq)
+ HCO
3-(aq)
= CH
3
-CHOH-COO
-(aq)
+ H
2
CO
3(aq)
Etat initial : 0,003 0,027 0 0,0014 en mol.L
-1
Etat final : ε 0,024 0,003 0,0044
Après avoir pris en compte cette réaction, la solution contient A
-
, HCO
3-
et H
2
CO
3
. La réaction
prépondérante n°2 est un équilibre de contrôle qui ne modifie pas les concentrations des espèces
présentes : H
2
CO
3(aq)
+ HCO
3-(aq)
= HCO
3-(aq)
+ H
2
CO
3(aq)
On peut alors obtenir le pH : pH = pK
a1
+ log[HCO
3-
]/[H
2
CO
3
] = 7,1.
6) Ce pH est en fait régulé par les concentrations des espèces carbonées, qui dépendent du rythme des
inspirations/expirations. Ici, pour voir remonter le pH vers sa valeur initiale, il faut une diminution de
H
2
CO
3
et donc de fortes expirations de gaz permettant une importante évacuation du dioxyde de
carbone.

PC A - PC B CHIMIE - DS n°5 - Correction
4
Partie 3 :
pH de mélanges
1) Calculons le pH d’un mélange de 20 mL d’acide acétique 0,10 mol.L
-1
et de 10 mL d’acide
nitreux 0,20 mol.L
-1
. On commence par représenter sur une échelle de pK
A
les différentes
espèces présentes et les couples acido-basiques correspondants :
H
3
O+
pK
A
H
2
O
CH
3
COO
-
0
4,75
CH
3
COOH
HO
-
14
H
2
O
HNO
2(aq)
+ H
2
O
(l)
= H
3
O
+(aq)
+ NO
2-(aq)
C'est la réaction entre la base la plus forte et l'acide
le plus fort: K°
1
= 10
-3,6
.
Dans le milieu sont présents 3 acides et 1 base, 3 réactions sont
donc envisageables:
H
2
O
(l)
+ H
2
O
(l)
= HO
-(aq)
+ H
3
O
+(aq)
K°
3
= Ke = 10
-14
.
(1)
(2)
NO
2-
3,6
HNO
2
CH
3
COOH
(aq)
+ H
2
O
(l)
= CH
3
COO
-(aq)
+ H
3
O
+(aq)
K°
2
= 10
-4,75
.
(3)
On suppose que (1) est la réaction prépondérante (bien que
K°
1
et K°
2
soient assez proches), on verra au moment des
vérifications si cela était approprié).
(1) n'est pas quantitative, c'est un équilibre de contrôle.
De plus K°
1
<< 1 donc (1) est peu avancé.
K°
1
= [H
3
O
+
][NO
2-
]/[HNO
2
] = x
1
²/(C - x
1
)
≈
x
1
²/C (équilibre peu avancé)
Dont on déduit x
1
= [H
3
O
+
] = 4,0.10
-3
mol.L
-1
avec C = 0,2/3 (ne pas oublier la dilution !)
Enfin on obtient
pH = 2,4
.
Il faut vérifier les approximations faites :
Première approximation
(équilibre (1) = réaction prépondérante) :
Deuxième équilibre : à pH = 2,4, CH
3
COOH est majoritaire donc [CH
3
COOH]
≈
0,1.2/3
mol.L
-1
. K°
2
= [CH
3
COO
-
][H
3
O
+
]/[CH
3
COOH] = x
2
.10
-2,4
/6,6.10
-2
= 10
-4,75
d’où x
2
=
3,0.10
-4
mol.L
-1
. On trouve 10.x
2
< x
1
donc l’équilibre (2) est bien négligeable devant (1).
Remarque : si on écrit K°
2
= [CH
3
COO
-
][H
3
O
+
]/[CH
3
COOH] = x
2
.(x
1
+ x
2
)/6,6.10
-2
, on
trouve x
2
= 2,8.10
-4
mol.L
-1
, on aboutit à un résultat similaire.
Troisième équilibre : on a pH
≤
6,5 donc on peut négliger l’autoprotolyse de l’eau.
Deuxième approximation
(équilibre (1) peu avancé) :

PC A - PC B CHIMIE - DS n°5 - Correction
5
pK
A
pH
pK
A
- 1 pK
A
+ 1
HNO
2
NO
2-
2,6
3,6
4,6
2,4 A pH = 2,4 on est bien dans le domaine où HNO
2
est majoritaire, la deuxième approximation est donc
validée, la valeur du pH également.
2) Calculer le pH d’un mélange NH
4
Cl 0,1 mol.L
-1
+ NH
4
CH
3
COO 0,05 mol.L
-1
.
H
3
O+
pK
A
H
2
O
CH
3
COO
-
0
4,75
CH
3
COOH
HO
-
14
H
2
O
NH
4+(aq)
+ CH
3
COO
-(aq)
= NH
3(aq)
+ CH
3
COOH
(aq)
C'est la réaction entre la base la plus forte et l'acide
le plus fort: K°
1
= 10
-4,5
.
Dans le milieu sont présents 2 acides et 2 bases, 4 réactions sont
donc envisageables:
H
2
O
(l)
+ H
2
O
(l)
= HO
-(aq)
+ H
3
O
+(aq)
K°
4
= K
e
= 10
-14
.
(1)
(2)
NH
3
9,25
NH
4+
CH
3
COO
-(aq)
+ H
2
O
(l)
= CH
3
COOH
(aq)
+ HO
-(aq)
K°
2
= 10
-9,25
.
(3)
(1) est la réaction prépondérante car K°
1
est beaucoup plus grande que les autres constantes
d'équilibre.
(4)
NH
4+(aq)
+ H
2
O
(l)
= NH
3(aq)
+ H
3
O
+(aq)
K°
3
= 10
-9,25
.
Si on réalise le tableau d’avancement correspondant à cette réaction :
NH
4+(aq)
+ CH
3
COO
-(aq)
= NH
3(aq)
+ CH
3
COOH
(aq)
Etat initial 0,15 0,05 0 0 en mol.L
-1
Etat équilibre 0,15 - x
1
0,05 - x
1
x
1
x
1
On a K°
1
= 10
-4,5
= x
1
²/((0,15 - x
1
)(0,05 - x
1
))
≈
x
1
²/(0,15.0,05) (puisque la réaction est peu
avancée). On en déduit
x
1
= 4,87.10
-4
mol.L
-1
.
Pour obtenir le pH, on utilise pH = pKa + log([A
-
]/[AH]) ou bien on effectue de produit
K
A(NH4+/NH3)
.K
A(CH3COOH/CH3COO-)
:
K
A(NH4+/NH3)
.K
A(CH3COOH/CH3COO-)
= [NH
3
][H
3
O
+
]/[NH
4+
].[CH
3
COO
-
][H
3
O
+
]/[CH
3
COOH]
soit K
A(NH4+/NH3)
.K
A(CH3COOH/CH3COO-)
= [CH
3
COO
-
][H
3
O
+
]²/[NH
4+
]
d’où [H
3
O
+
] = (10
-9,25
.10
-4,75
.0,15/0,05)
1/2
= 1,73.10
-7
mol.L
-1
soit
pH = 6,8
.
Il faut vérifier les approximations faites :
Première approximation
(équilibre (1) = réaction prépondérante) :
 6
7
8
9
10
11
12
13
14
15
16
17
18
6
7
8
9
10
11
12
13
14
15
16
17
18
1
/
18
100%