radiographiée radiographiée

Mensuel pour médecins, médecins-dentistes et pharmaciens
N°13 - FEVRIER 2010
PERINDOPRIL
ÉVASION FINANCE PEOPLE
ACTU
Nouvelles de l’association
pharma luxembourgeoise
Dr Henri Kugener:
un collectionneur passionné
Les impôts vont-ils augmenter?
Il est cinq heures,
Bangkok s’endort
L’EXPERT DU MOIS
Dr Remy Demuth
La GASTRO
radiographiée
La GASTRO
radiographiée

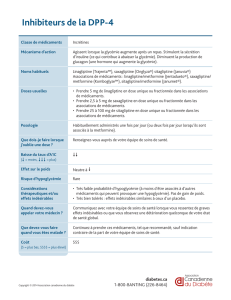
1. DENOMINATION DU MEDICAMENT: Janumet 50 mg/850 mg, comprimés pelliculés. Janumet 50 mg/1000 mg, comprimés pelli-
culés. 2. COMPOSITION QUALITATIVE ET QUANTITATIVE: Chaque comprimé contient 50 mg de sitagliptine (sous forme de phosphate
monohydraté) et 850 mg de chlorhydrate de metformine. Chaque comprimé contient 50 mg de sitagliptine (sous forme de phosphate
monohydraté) et 1000 mg de chlorhydrate de metformine. 3. FORME PHARMACEUTIQUE: Comprimé pelliculé (comprimé). Janumet
50 mg/850 mg: Comprimé pelliculé en forme de gélule, de couleur rose portant l’inscription “515” sur une face. Janumet 50 mg/1000
mg: Comprimé pelliculé en forme de gélule, de couleur rouge portant l’inscription “577” sur une face. 4. DONNEES CLINIQUES:
4.1 Indications thérapeutiques: Chez les patients diabétiques de type 2, Janumet est indiqué pour améliorer le contrôle de la glycémie, en
dose maximale tolérée ou chez les patients déjà traités par l’association sitagliptine/metformine. s en association à un sulfamide hypo-
glycémiant (trithérapie) lorsque les doses maximales tolérées de metformine et de sulfamide ne permettent pas d’obtenir un contrôle adé-
quat de la glycémie. s en trithérapie avec un agoniste des récepteurs PPARγ (thiazolidinedione) lorsque les doses maximales tolérées de
metformine et de l’agoniste des récepteurs PPARγ ne permettent pas d’obtenir un contrôle adéquat de la glycémie. 4.2 Posologie et mode
d’administration: La posologie du traitement antihyperglycémiant par Janumet doit être adaptée au patient en fonction de son traitement
contrôlés par la metformine en monothérapie, la dose initiale habituelle de Janumet doit être: sitagliptine à raison de 50 mg deux fois par
jour (dose quotidienne totale de 100 mg) plus metformine à la posologie déjà prise par le patient. Patients déjà traités par la sitagliptine
et la metformine en association (substitution). Chez
les patients qui prenaient la sitagliptine et la met-
formine sous forme de comprimés séparés, le trait-
ement par Janumet doit être instauré aux doses de
sitagliptine et de metformine déjà prises par le
une bithérapie metformine/sulfamide hypoglycémi-
ant aux doses maximales tolérées. La posologie de
Janumet doit apporter 50 mg de sitagliptine deux
fois par jour (dose quotidienne totale de 100 mg)
et une dose de metformine égale à la dose déjà
prise. Lorsque Janumet est utilisé en association
à un sulfamide hypoglycémiant, une réduction de
la posologie du sulfamide hypoglycémiant peut être
envisagée pour réduire le risque d’hypoglycémie.
bithérapie metformine et agoniste des récepteurs
PPARγ aux doses maximales tolérées. La posologie
de Janumet doit apporter 50 mg de sitagliptine
deux fois par jour (dose quotidienne totale de 100
mg) et une dose de metformine égale à la dose déjà
prise par le patient. Pour permettre les différentes
posologies de metformine, Janumet est disponible
aux dosages de 50 mg de sitagliptine et 850 mg
ou 1000 mg de chlorhydrate de metformine. Tous
les patients doivent poursuivre leur régime ali-
mentaire, avec une répartition régulière de l’apport
glucidique au cours de la journée. Les patients en
surpoids doivent poursuivre leur régime hypocalo-
rique. Janumet doit être pris deux fois par jour au
cours des repas pour diminuer les effets indésir-
ables gastro-intestinaux associés à la metformine.
être utilisé chez les patients présentant une insuf-
créatinine <60 ml/mn) (voir rubrique 4.3). Patients
utilisé chez les patients présentant une insuf-
La metformine et la sitagliptine étant éliminées par
voie urinaire, Janumet doit être administré avec
prudence chez les patients âgés. La fonction rénale
devra être surveillée pour prévenir une acidose lac-
tique associée à la metformine, en particulier chez
les sujets âgés (voir rubrique 4.3). On dispose de
données limitées de tolérance pour la sitagliptine
chez les patients âgés de plus de 75 ans. La pru-
dence est donc requise. Enfants: Compte tenu de
Janumet n’est pas recommandé chez l’enfant de
moins de 18 ans. 4.3 Contre-indications: Janumet
est contre-indiqué chez les patients avec: - hy-
persensibilité aux substances actives ou à l’un
des excipients (voir rubrique 4.8); - acidocétose
rénale modérée ou sévère (clairance de la créati-
nine <60 ml/mn); - affections aiguës susceptibles
d’altérer la fonction rénale, tels que: - déshydra-
tation, - infection grave, - choc, - administration
intravasculaire de produits de contraste iodés; -
maladies aiguës ou chroniques pouvant provoquer
cardiaque ou respiratoire, - infarctus du myocarde
-
cation éthylique aiguë, alcoolisme; - allaitement.
4.8 Effets indésirables: Aucun essai thérapeutique
n’a été mené avec Janumet comprimés, mais la
bioéquivalence de Janumet avec la sitagliptine et
la metformine co-administrées a été démontrée.
Sitagliptine et metformine: Les effets indésirables
considérés comme imputables au médicament et
rapportés avec une plus grande fréquence (>0,2%
et différence >1 patient) chez les patients recevant
la sitagliptine en association avec la metformine
que chez ceux sous placebo, dans les études en
double aveugle, sont répertoriés ci-après selon les
termes MedDRA, par classes d’organe et fréquences
r1/10); fréquent (r1/100, <1/10); peu fréquent (r1/1000,
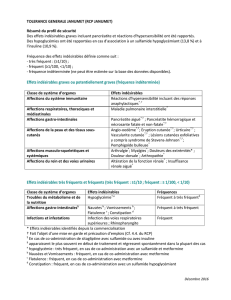
<1/100); rare (r1/10000, <1/1000); et très rare (<1/10000). Tableau 1. Fréquence des effets indésirables dans les études cliniques
versus placebo.
Effets indésirables Fréquence des effets indésirables par traitement
sitagliptine
+metformine 1
sitagliptine
+ metformine et sulfamide
hypoglycémiant 2
sitagliptine +
metformine et agoniste
des récepteurs PPARγ
(rosiglitazone) 3
Temps de l’analyse Semaine 24 Semaine 24 Semaine 18
Investigations
baisse de la glycémie Peu fréquent
Affections du système nerveux
céphalées Fréquent
somnolence Peu fréquent
Affections gastro-intestinales
diarrhée Peu fréquent Fréquent
nausées Fréquent
constipation Fréquent
douleur abdominale haute Peu fréquent
Vomissements Fréquent
Troubles du métabolisme et de la nutrition
hypoglycémie* Très fréquent Fréquent
Troubles généraux
Œdème périphérique Fréquent†
* Dans les essais cliniques réalisés soit avec la sitagliptine en monothérapie, soit avec la sitagliptine en association à la metformine ou
à la metformine et agoniste des récepteurs PPARγ, l’incidence des hypoglycémies observées avec la sitagliptine était similaire à celle
des patients sous placebo. † Observé dans l’analyse à 54 semaines. 1Dans cette étude de 24 semaines, versus placebo, menée avec 100
mg de sitagliptine, une fois par jour, en ajout au traitement en cours par
la metformine, l’incidence des effets indésirables considérés comme im-
putables au médicament chez les patients traités par la sitagliptine en ajout
au traitement en cours par la metformine et chez les patients recevant le
placebo en ajout au traitement en cours par la metformine a été respective-
ment de 9,3% et 10,1%. Dans une étude supplémentaire d’un an réalisée
avec 100 mg de sitagliptine, une fois par jour, en ajout au traitement en
cours par la metformine, l’incidence des effets indésirables considérés comme imputables au médicament chez les patients traités par
la sitagliptine en ajout au traitement en cours par la metformine et chez les patients recevant un sulfamide hypoglycémiant en ajout
au traitement en cours par la metformine a été respectivement de 14,5% et 30,3%. Dans les études regroupées d’une durée allant
jusqu’à un an, comparant la sitagliptine en ajout au traitement en cours par la metformine à un sulfamide hypoglycémiant en ajout au
traitement en cours par la metformine, les effets indésirables considérés comme imputables au médicament et rapportés avec une plus
grande fréquence (>0,2% et différence >1 patient) chez les patients traités par 100 mg de sitagliptine que chez les patients traités
par le sulfamide hypoglycémiant sont les suivants : anorexie (Troubles du métabolisme et de la nutrition; peu fréquent) et réduction du
poids (Investigations; peu fréquent). 2Dans cette étude de 24 semaines versus placebo réalisée avec 100 mg de sitagliptine, une fois
par jour, en ajout au traitement en cours par l’association de glimépiride et de metformine, l’incidence globale des effets indésirables
considérés comme imputables au médicament a été de 18,1% chez les patients recevant la sitagliptine en plus de l’association gli-
mépiride/metformine et de 7,1% chez les patients
recevant le placebo en plus de l’association gli-
mépiride/metformine. 3Dans cette étude réalisée
pendant 54 semaines avec 100 mg de sitagliptine,
une fois par jour, en association à la rosiglitazone et
à la metformine, l’incidence des effets indésirables
considérés comme imputables au médicament, a
été respectivement de 15,3% et 10,9% chez les
patients traités par la sitagliptine en association
et chez ceux traités par placebo en association. Une
analyse à 54 semaines réalisée chez les patients
traités par la sitagliptine plus association par rap-
port à ceux traités uniquement par l’association a
montré que d’autres effets indésirables (fréquents)
et imputables au traitement ont été rapportés avec
une plus grande fréquence (>0,2% et différence >1
patient): céphalées, toux, vomissements, hypogly-
cémie, infection fongique cutanée et infection des
voies respiratoires supérieures. Dans une étude de
24 semaines réalisée avec l’association sitaglip-
tine/metformine, en traitement initial, administrée
2 fois par jour (sitagliptine/metformine 50 mg/500
mg ou 50 mg/1000 mg), l’incidence globale des
effets indésirables considérés comme imputables
au médicament a été respectivement de 14,0%
et 9,7% chez les patients traités par l’association
sitagliptine/metformine et chez les patients sous
placebo. L’incidence globale des effets indésirables
considérés comme imputables au médicament chez
les patients traités par l’association sitagliptine/
metformine a été comparable à celle des patients
traités par metformine seule (14,0% dans chaque
groupe) et supérieure à celle des patients traités
par sitagliptine seule (6,7%), les différences par
rapport à la sitagliptine seule étant principalement
dues aux effets indésirables gastro-intestinaux.
Autres informations sur chacune des substances
dans des études en monothérapie d’une durée al-
lant jusqu’à 24 semaines, réalisées avec 100 mg de
sitagliptine, une fois par jour, versus placebo, les
effets indésirables considérés comme imputables
au médicament et rapportés avec une plus grande
fréquence (>0,2% et différence >1 patient) chez
les patients traités par la sitagliptine que chez
les patients sous placebo sont les céphalées,
l’hypoglycémie, la constipation et les étourdisse-
ments. En plus des effets indésirables imputables
au médicament décrits ci-dessus, des effets indé-
sirables ont été rapportés indépendamment de la
relation de cause à effet avec le médicament chez
au moins 5% des patients, et plus fréquemment
chez ceux traités par sitagliptine. Ils comprenaient
des infections des voies respiratoires supérieures
et des rhino-pharyngites. D’autres événements
indésirables sont survenus plus fréquemment
chez les patients traités par sitagliptine (sans
atteindre le seuil de 5%, mais avec une incidence
supérieure chez les patients traités par sitagliptine
de plus de 0,5% par rapport au groupe contrôle). Ils
comprenaient arthrose et douleurs des extrémités.
Dans l’ensemble des études cliniques, une légère
augmentation du nombre de glo-bules blancs (dif-
férence d’environ 200 globules blancs/microlitre
versus placebo; valeur moyenne de départ d’environ
6600 globules blancs/microlitre) a été observée, en
raison d’une augmentation du nombre de neutro-
philes. Cette observation a été constatée dans la
plupart des études mais pas dans toutes. Cette
considérée comme cliniquement pertinente. Aucune
vitaux ou de l’ECG (y compris l’intervalle QTc) n’a
été observée avec le traitement par sitagliptine.
Données de pharmacovigilance: Depuis la com-
mercialisation de Janumet ou de la sitagliptine,
une des substances actives de Janumet, les effets
indésirables supplémentaires suivants ont été rapportés (fréquence indéterminée): réactions d’hypersensibilité incluant anaphylaxie,
angio-œdème, rash, urticaire, vascularite cutanée et lésions cutanées exfoliatives y compris syndrome de Stevens-Johnson; pancréatite.
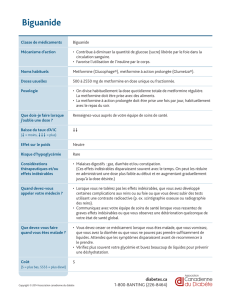
Metformine: Données des études cliniques et données de pharmacovigilance. Le tableau 2 présente les effets indésirables par classe
Produit de la metformine disponible dans l’Union européenne. Tableau 2. Fréquence des effets indésirables de la metformine à partir
des données des études cliniques et de la pharmacovigilance.
Effets indésirables Fréquence
Affections du système nerveux
goût métallique Fréquent
Affections gastro-intestinales
symptômes gastro-intestinauxa Très fréquent
Affections de la peau et du tissu sous-cutané
urticaire, érythème, prurit Très rare
Troubles du métabolisme et de la nutrition
acidose lactique Très rare
carence en vitamine B12bTrès rare
Affections hépatobiliaires
Troubles de la fonction hépatique, hépatite Très rare
a Les symptômes gastro-intestinaux tels que nausées, vomissements, diarrhées, douleurs abdominales et perte d’appétit appa-
raissent le plus souvent en début de traitement et régressent spontanément dans la plupart des cas. b Le traitement au long cours
par la metformine a été associé à une diminution de l’absorption de la vitamine B12 qui peut, dans de très rares cas, entraîner
7. TITULAIRE DE L’AUTORISATION
DE MISE SUR LE MARCHE: Merck Sharp & Dohme Ltd. - Hertford Road, Hoddesdon - Hertfordshire EN11 9BU - Royaume-Uni.
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE: EU/1/08/455/001 - EU/1/08/455/002 - EU/1/08/455/003 - EU/1/08/455/004
- EU/1/08/455/005 - EU/1/08/455/006 - EU/1/08/455/007 - EU/1/08/455/015 - EU/1/08/455/008 - EU/1/08/455/009 - EU/1/08/455/010
- EU/1/08/455/011 - EU/1/08/455/012 - EU/1/08/455/013 - EU/1/08/455/014 - EU/1/08/455/016. 9. DATE DE PREMIERE AUTORISATION/
DE RENOUVELLEMENT DE L’AUTORISATION: 16 juillet 2008. 10. DATE DE MISE A JOUR DU TEXTE: 16 septembre 2009. Mode de délivrance:
sur prescription médicale.
(sitagliptine/metformine, MSD)
Nouveau
Changing the course to glucose control.
®
Dès à présent disponible
® Registered Trademark of Merck Sharp & Dohme Corp.,
a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, U.S.A.
All rights reserved. 31-Dec-2010 JMT-2009-BE-2470-J.
1135, Chaussée de Waterloo - 1180 Bruxelles.
L’illustration est une interprétation artistique.
Non représentatif des effets cliniques.
JANUMET 50/850 mg et 50/1000 mg
56 comp. = 51.72 €
196 comp. = 157.97 €

3
ÉDITO
Rédacteur en chef
Dr Eric Mertens
Secrétaire de rédaction
Françoise Moitroux
Directrice artistique
Nathalie Ruykens
Ont collaboré à ce numéro
Dr J. Andris, F. Anselme, S. Calmant,
O. Frochisse, Pierre Kroll,
Dr M. Langendries, Dr Ch. Maton
Dr J.L. Schouveller, Pr E. Van Cutsem,
M. Vandermeir, Dr J. Vannoote.
Photographe Semper
Luc Deflorenne
www.lucphoto.lu
Production
Sacha Design s.à.r.l.
Impression
Saint-Paul Luxembourg s.a.
www.saint-paul.lu
Editeur
Semper Luxembourg est une publication
de DSB Communication sous
licence Roularta Media Group
DSB Communication s.a.
Société anonyme au capital de 31.000 €
Adm. responsable : Dr Corinne Rosman
31 Val Sainte Croix - L-1371 Luxembourg
Tél. +352 26 25 61 41 - Fax +352 26 25 82 01
R.C.S. Luxembourg B 110.223
Autorisation d’établissement N°123743
Directeur général
Dr Eric Mertens
Les articles, photos, dessins et autres illustrations de la partie rédactionnelle de Semper ne comportent pas de publicité. Les mentions d’entreprises ou de produits figurent à titre
documentaire. Les articles, photos et dessins ainsi que les opinions et les publicités sont publiés sous la seule responsabilité des auteurs ou annonceurs. Tous droits de traduction,
d’adaptation et de reproduction, par quelque procédé que ce soit, sont réservés pour tous pays.
Après les
plombiers
polonais…
C’est en 2005 – déjà ! – que le “plombier polonais” est entré dans le langage cou-
rant. Née en France à l’occasion du débat sur le traité constitutionnel européen, l’ex-
pression a été fortement médiatisée, et est devenue le symbole du dumping social.
Dans la même veine, mais plus lointains, sont apparus les “ingénieurs indiens”, autre
main d’oeuvre à bas prix à l’origine d’une concurrence déloyale. Dans ce numéro de
Semper Luxembourg, le Dr Demuth nous apprend que l’Inde se profile également
dans le champ d’action de la radiologie. Voilà qui est plus fâcheux que le plombier
polonais, car la transformation d’un acte médical en un service commercial n’augure
rien de bon.
Mais il y a pire encore que le radiologue indien, souffle-t-on dans les cercles médi-
caux: c’est le “médecin allemand”. Ah, cette fois on ne parle plus de bradage des
prix. Ce serait même plutôt le contraire.
Que l’on s’entende: loin de nous l’idée d’embrasser un quelconque antigermanisme,
ou de mettre en cause la médecine du grand voisin. Non, le défaut de ce confrè-
re d’un genre nouveau, qui peut d’ailleurs tout aussi bien être français, belge ou
moldo-valaque (bien que ce soit plus rare), ne tient pas à sa nationalité. Mais bien à
son choix d’entreprise.
Car ouvrir une consultation un ou deux jours par semaine au Grand-Duché pour
compléter un horaire incomplet à Trèves ou ailleurs, ce n’est pas une approche médi-
cale orientée vers le patient, mais bien une démarche affairiste. Un choix fort éloigné
du suivi et de la disponibilité que le patient luxembourgeois est en droit d’attendre.
Voilà ce qui est fâcheux, nous dites-vous…
Dr Eric Mertens

Il n’y a pas que
le cancer du col
de l’utérus…
ENSEMBLE avec GARDASIL®,
nous pouvons faire plus…
Prévention du cancer du col de l’utérus
et aussi :
• des lésions génitales précancéreuses
(du col de l’utérus, du vagin, de la vulve)
• des verrues génitales externes
liées aux 4 types d’HPV 6,11,16 et 18
REMBOURSE POUR TOUTES LES
JEUNES FILLES DE 12 à 18 ANS
p.p. � 123,44
DENOMINATION DU MEDICAMENT Gardasil, suspension injectable en seringue préremplie. Vaccin
Papillomavirus Humain [Types 6, 11, 16, 18] (Recombinant, adsorbé) COMPOSITION QUALITATIVE
ET QUANTITATIVE 1 dose (0,5 ml) contient environ: Protéine L1 de Papillomavirus Humain1 de
type 62, 3 20 microgrammes Protéine L1 de Papillomavirus Humain1 de type 112, 3 40 micro-
grammes Protéine L1 de Papillomavirus Humain1 de type 162, 3 40 microgrammes Protéine L1 de
Papillomavirus Humain1 de type 182, 3 20 microgrammes 1 Papillomavirus Humain = HPV. 2 Protéine
L1 sous la forme de pseudo particules virales produite sur des cellules de levure (Saccharomyces
cerevisiae CANADE 3C-5 (souche 1895)) par la technique de l’ADN recombinant. 3 adsorbée sur
sulfate d’hydroxyphosphate d’aluminium amorphe (Al: 225 microgrammes) comme adjuvant.
FORME PHARMACEUTIQUE Suspension injectable en seringue préremplie. Avant agitation, Gardasil
peut apparaître comme un liquide clair avec un précipité blanc. Après une agitation minutieuse, le li-
quide est blanc, trouble. INDICATIONS THÉRAPEUTIQUES Gardasil est un vaccin pour la prévention
des lésions génitales précancéreuses (du col de l’utérus, de la vulve et du vagin), des cancers du col
de l’utérus et des verrues
génitales externes (condy-
lomes acuminés) dus aux
Papillomavirus Humains
(HPV) de types 6, 11, 16 et
18. L’indication est fondée
sur la démonstration de
l’effi cacité de Gardasil chez
les femmes adultes de
16 à 26 ans et sur la dé-
monstration de l’immuno-
génicité de Gardasil chez
les enfants et adolescents
de 9 à 15 ans. L’effi cacité
protectrice n’a pas été éva-
luée chez les sujets de sexe
masculin. Gardasil doit être
utilisé sur la base des re-
commandations offi cielles.
POSOLOGIE ET MODE
D’ADMINISTRATION Le
schéma de primovacci-
nation comprend 3 doses
de 0,5 ml administrées
selon le schéma suivant :
0, 2, 6 mois. Si un autre
schéma de vaccination
s’avère nécessaire, la deu-
xième dose doit être admi-
nistrée au moins un mois
après la première dose,
et la troisième dose doit
être administrée au moins
3 mois après la deuxième
dose. Les trois doses doi-
vent être administrées en
moins d’un an. La nécessi-
té d’une dose de rappel n’a
pas été établie. Population
pédiatrique: Gardasil n’a
pas été utilisé chez les en-
fants de moins de 9 ans. Le
vaccin doit être administré
par voie intramusculaire.
Le vaccin doit être injecté
de préférence dans la
région deltoïdienne de la
partie supérieure du bras
ou dans la région antéro-
latérale supérieure de la
cuisse. Gardasil ne doit
pas être injecté par voie
intravasculaire. Les admi-
nistrations sous-cutanée
et intradermique n’ont pas
été évaluées. Ces modes
d’administration ne sont
pas recommandées. Il est
recommandé aux sujets qui
ont reçu une première dose
de Gardasil de terminer
le schéma de vaccination
en 3 doses avec Gardasil.
CONTRE-INDICATIONS
Hypersensibilité aux substances actives ou à l’un des excipients du vaccin. Les sujets ayant présenté
des symptômes indiquant une hypersensibilité après l’administration d’une dose de Gardasil ne doi-
vent pas recevoir d’autres doses de Gardasil. L’administration de Gardasil doit être différée chez les
individus souffrant d’une maladie fébrile aiguë sévère. Cependant, la présence d’une infection mi-
neure, comme une infection modérée des voies respiratoires supérieures ou une fi èvre peu élevée,
n’est pas une contre-indication à la vaccination. EFFETS INDÉSIRABLES Dans 6 études cliniques
(dont 5 contrôlées contre placebo), les sujets ont reçu Gardasil ou le placebo le jour de leur inclusion
et approximativement 2 mois et 6 mois plus tard. Peu de sujets (0,2%) sont sortis d’étude en raison
d’effets indésirables. La tolérance a été évaluée soit sur toute la population de l’étude (4 études)
soit sur un sous-groupe prédéfi ni de la population de l’étude (1 étude) en utilisant
des carnets de surveillance pendant 14 jours après chaque injection de Gardasil ou
du placebo. 8 068 sujets ayant reçu Gardasil (6 996 femmes âgées de 9 à 45 ans
et 1072 garçons âgés de 9 à 15 ans lors de l’inclusion) et 5 966 sujets ayant reçu
le placebo ont été suivis à l’aide de carnets de surveillance. Chez
les sujets ayant reçu Gardasil, les effets indésirables liés au vaccin,
mentionés ci-dessous, ont été observés soit à une fréquence d’au
moins 1%, soit à une fréquence plus importante que celle qui a été
observée chez les sujets ayant reçu le placebo. Ils sont classés en fonction de leur fréquence selon
la convention suivante: [Très fréquent (≥ 1/10), Fréquent (≥ 1/100, <1/10), Peu fréquent (≥ 1/1 000,
< 1/100), Rare (≥ 1/10 000, < 1/1 000, Très rare (<1/10 000), y compris cas isolés]
Troubles musquolo-squelettiques et du tissu conjonctif Fréquent: douleur des extrêmités.
Troubles généraux et anomalies liées au site d’administration : Très fréquent : fi èvre. Très
fréquent: au site d’injection : érythème, douleur, gonfl ement. Fréquent : au site d’injection :
ecchymose, prurit. De plus, au cours des études cliniques, des effets indésirables qui ont
été jugés par l’investigateur en relation avec le vaccin ou avec le placebo, ont été observés
à des fréquences inférieures à 1%: Affections respiratoires, thoraciques et médiastinales :
Très rare : bronchospasme.
Affections de la peau
et du tissu sous-cutané
: Rare : urticaire. Neuf
cas (0,07%) d’urticaire
ont été rapportés dans le
groupe Gardasil et 16 cas
(0,18%) dans le groupe
placebo contenant l’ad-
juvant. Dans les études
cliniques, les sujets faisant
l’objet d’un suivi de tolé-
rance ont rapporté tous
les nouveaux événements
médicaux pendant une pé-
riode allant jusqu’à 4 ans.
Parmi les 13 686 sujets
ayant reçu Gardasil et
les 11 588 sujets ayant
reçu le placebo, 35 cas
d’arthrites non spécifi ques
ont été rapportés, 20 dans
le groupe Gardasil et
13 dans le groupe placebo.
Dans une étude clinique
conduite chez 843 ado-
lescents, fi lles et garçons
âgés de 11 à 17 ans,
l’administration conco-
mitante de la première
dose de Gardasil avec un
vaccin combiné de rappel
diphtérique, tétanique, co-
quelucheux [acellulaire] et
poliomyélitique [inactivé]
a montré qu’il y avait plus
de gonfl ements au site
d’injection et de céphalées
rapportés suite à l’admi-
nistration concomitante.
Les différences observées
étaient < 10% et chez la
majorité des sujets, les
événements indésirables
étaient rapportés avec une
intensité faible à modérée.
Expérience après mise
sur le marché Des événe-
ments indésirables ont été
spontanément rapportés
après la mise sur le marché
de Gardasil et ne sont pas
listés ci-dessus : Comme
ces évènements ont été
rapportés volontairement
à partir d’une population
de taille incertaine, il n’est
pas possible d’estimer de
manière fi able leur fré-
quence ni d’établir, pour
tous ces événements, un
lien de cause à effet avec
la vaccination. Affections
hématologiques et du
système lymphatique: adénopathie. Troubles du système immunitaire: réactions d’hypersensibilité
incluant des réactions anaphylactiques/anaphylactoïdes. Troubles du système nerveux : syndrome
de Guillain-Barré, sensation de vertige, céphalée, syncope parfois accompagnée de mouvements
tonico-cloniques. Troubles gastro-intestinaux: nausées, vomissements. Troubles musculosquelet-
tiques, du tissu conjonctif et des os : arthralgie, myalgie. Troubles généraux et anomalies liés au site
d’administration : asthénie, frissons, fatigue, malaise. NATURE ET CONTENU DE L’EMBALLAGE
EXTÉRIEUR 0,5 ml de suspension en seringue préremplie (verre de type I) munie d’un bouchon-
piston (élastomère bromobutyl recouvert de FluoroTec siliconé ou élastomère chlorobutyl) et d’un
capuchon (bromobutyl) sans dispositif de protection de l’aiguille, avec deux aiguilles – boîtes de 1.
TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE Sanofi Pasteur MSD
SNC, 8 rue Jonas Salk, F-69007 Lyon, France NUMERO D’AUTORISATION DE MISE
SUR LE MARCHE EU/1/06/357/007 DELIVRANCE Médicament soumis à prescrip-
tion médicale DATE DE MISE A JOUR DU TEXTE 2 septembre 2009 BE00173/10-2009

Semper Luxembourg - février 2010
N°13 - FEVRIER 2010 Dans ce
numéro
Histoire de la médecine
Au temps du silex chirurgical
Le Dr Henri Kugener (dont vous pouvez
découvrir le portrait dans notre rubrique
PEOPLE) nous commente l’impressionnant
savoir-faire chirurgical de nos ancêtres.
Dossier médical
La gastro radiographiée
Reflux gastro-oesophagien, maladie de
Crohn, rectocolite, cancer du côlon...
Le dossier du mois fait le point, avec en
conclusion le regard du Dr Remy Demuth,
qui nous apporte son éclairage sur la place
mouvante de l’imagerie en gastro-entéro-
logie et dans le paysage luxembourgeois.
Finance
Les impôts vont-ils augmenter ?
Le gouvernement doit trouver de nou-
velles pistes pour faire rentrer de l’argent
dans les caisses. L’une des pistes serait une
augmentation des impôts, plus rien ne
serait donc tabou pour la classe politique
luxembourgeoise.
Évasion
Il est cinq heures,
Bangkok s’endort
Quelques conseils pour apprivoiser cette
capitale trépidante et affolante: découvrez
les bateaux-bus, le Grand Palais, les mar-
chés spectaculaires sans oublier la surpre-
nante gastronomie thaïe…
Actualité
Nouvelles de l’association
pharma luxembourgeoise 06
Flash
News du médicament 08
People
Dr Henri Kugener:
rencontre avec un
collectionneur passionné 10
Histoire de la médecine
Au temps du silex chirurgical 13
Dossier médical
l
Reflux gastro-oesophagien,
le grand retour 16
l
Crohn, l’opacité se lève 20
l
Intestins et articulations,
unis par la pathologie 24
l
Cancer du côlon: quelles
stratégies de prévention
et de dépistage ? 26
l
L’expert du mois:
Dr Remy Demuth 30
Finance
Les impôts vont-ils
augmenter ? 34
Evasion
La glisse chic 36
Thaïlande: capitale Bangkok 38
On en parle
De Yasmina Khadra
à Quentin Tarantino 40
Agenda 41
Le meilleur de Kroll
Biologie de l’homosexualité 42
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
1
/
44
100%