D`une manière générale, l`inhabituel jeune âge au

Prédisposition au cancer du sein
Dr Olivier Caron
Gustave Roussy, Villejuif
Mars 2015
Objectifs :
- Savoir reconnaître les indications de consultation de génétique dans les
cancers du sein
- Connaître les modalités de prise en charge des personnes porteuses de
mutation BRCA1 ou 2.
- Connaître l’impact d’une mutation constitutionnelle sur la prise en charge
thérapeutique
Le cancer résulte de la conjonction de 3 facteurs : l’environnement, le
patrimoine génétique, le hasard. Il est important de distinguer patrimoine génétique
constitutionnel d’une personne, présent dans toutes les cellules nucléées d’un
individu, et matériel génétique somatique, qui ne concerne que certaines cellules,
notamment tumorales. La mutation du patrimoine génétique constitutionnel entraîne
la survenue d’une prédisposition.
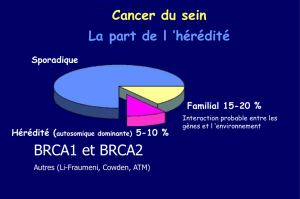
Plusieurs gènes peuvent, lorsqu’ils sont porteurs d’anomalies génétiques, être
responsables d’une augmentation majeure du risque de cancer du sein. Deux
éléments permettent de suspecter une anomalie constitutionnelle: le nombre
inhabituel de cancer du sein dans une famille, et l’âge précoce des cancers. Les
situations suivantes doivent mener à la réalisation d’une consultation de génétique et
constituent des indications d’analyse de BRCA1 et BRCA2 :
- au moins 3 cas de cancers du sein chez des apparentées proches
- au moins deux cas, dont un diagnostiqué avant l’âge de 45 ans ou un cas
masculin
- un seul cas de cancer du sein, diagnostiqué avant 35 ans
- l’association chez une même personne ou dans une même famille d’un cancer
du sein et d’un cancer de l’ovaire.
- Un seul cas de cancer de l’ovaire de haut grade (séreux+++), diagnostiqué
avant 70 ans
- Fréquemment admis, mais non encore consensuel : un seul cas de cancer du
sein triple négatif, diagnostiqué avant 50 ans
1

La première étape consiste à identifier une anomalie dans une famille où elle est
suspectée. A ce niveau, une seule analyse est menée par famille, chez la personne
chez laquelle la probabilité de trouver l’anomalie si elle existe est la plus grande. Il
s’agit de la personne ayant développé le cancer à l’âge le plus jeune.
La consultation d’oncogénétique doit être une démarche volontaire de la part du
patient. Lors de cet entretien, l’oncogénéticien précise la structure familiale et les
antécédents. En fonction de tous ces éléments, il valide l’indication d’analyse
génétique.
Le résultat de cette analyse révèlera :
- soit une mutation délétère (en moyenne, dans 10-15% des situations ci-
dessus), authentifiant la prédisposition
- soit aucune anomalie particulière
- soit un variant de signification inconnue.
Les deux derniers cas ne permettent pas d’exclure une prédisposition.
Risques associés aux mutations constitutionnelles de BRCA1 ou BRCA2
Chez la femme, une mutation de BRCA1 confère un risque de cancer du sein
d’environ 60-70% et un risque de cancer de l’ovaire de 20 à 40%. Une mutation de
BRCA2 génère un risque équivalent de cancer du sein, mais un risque de cancer de
l’ovaire significativement inférieur, de l’ordre de 10 à 20%.
2

Les cancers du sein associés à BRCA1 sont préférentiellement « triple-négatifs »
(absence de récepteurs hormonaux, absence de surexpression de cerbB2), mais tous
les cancers triple-négatifs ne correspondent pas à une mutation constitutionnelle de
BRCA1.
Les cancers de l’ovaire sont préférentiellement une histologie séreuse. Il ne s’agit
pratiquement jamais de forme mucineuse. Les tumeurs germinales de l’ovaire ne font
pas partie du spectre BRCA, pas plus que les tumeurs borderline de l’ovaire.
Chez l’homme, un risque de cancer du sein existe notamment avec BRCA2 mais est
nettement moindre (risque absolu de 2-4%). Un sur-risque de cancer de la prostate
de quelques pourcents est habituellement retenu.
Pour les deux sexes, un sur-risque de cancer du pancréas est également suspecté
pour une mutation de BRCA2.
Prise en charge des femmes porteuses de mutation BRCA1 et BCRA2
Prise en charge du risque de cancer du sein:
Il est recommandé de réaliser chez les personnes porteuses d’une mutation BRCA1
ou 2 :
- une surveillance clinique, dès l’âge de 25 ans, par palpation mammaire chez le
spécialiste 2 fois par an
- des examens d’imagerie : mammographie et échographie mammaire
annuelles dès l’âge de 30 ans. Les données récentes de la littérature
permettent d’ajouter la réalisation d’IRM mammaires systématiques. Il est
actuellement préconisé de réaliser les 3 examens simultanément. Cette
position est susceptible d’évoluer.
Il n’y a pas d’examen biologique de dépistage. Cette surveillance est mise en place
sans limite d’âge, ce qui exclut ces femmes du dépistage de masse.
La mastectomie prophylactique n’est pas recommandée systématiquement, la
surveillance étant mise en avant préférentiellement. Il s’agit d’une option dont la
patiente doit être systématiquement informée. Elle doit être accompagnée dans son
choix, éclairée par les conseils techniques notamment des chirurgiens et d’un soutien
psychologique. Un délai de réflexion suffisant doit être respecté. Il est variable selon
les personnes, en fonction des niveaux d’information et de réflexion préalables.
L’indication de tout geste prophylactique doit être validée de manière
collégiale en réunion de concertation pluridisciplinaire.
Prise en charge du risque de cancer de l’ovaire:
Aucune surveillance ovarienne ne permet de dégager de bénéfice médical. Il est
cependant d’usage de proposer une échographie pelvienne annuelle à partir de l’âge
de 35 ans. Le manque de sensibilité et de spécificité de cet examen fait porter la
recommandation d’une annexectomie prophylactique à la quarantaine. Ce geste
radical permet de diminuer considérablement le risque de cancer de l’ovaire (risque
résiduel : environ 2%), et de diminuer le risque de cancer du sein d’environ 50% si il
est effectué avant 45 ans.
3

Autres mesures :
La contraception ne fait l’objet d’aucune mesure particulière, malgré un léger
surrisque.
Un traitement hormonal substitutif de la ménopause est envisageable pour les
femmes ménopausée après annexectomie prophylactique à un âge jeune.
Mesures pour les apparentés :
Toute personne porteuse d’une mutation génétique peut à son tour transmettre
l’anomalie génétique, avec un risque de 50% à chaque grossesse, quel que soit le
sexe du parent ou celui de l’enfant. Les tests présymptomatiques peuvent alors être
réalisés chez les enfants majeurs après réalisation d’une consultation de génétique.
Pour les apparentées non porteuse : pas de prise en charge spécifique
Pour les porteuses : les recommandation ci-dessus s’appliquent.
Cas des familles évocatrices de prédisposition sans mutation identifiable :
Lorsqu’aucune mutation n’est identifiée dans une famille, une surveillance adaptée
est proposée aux apparentées au premier degré de femmes ayant développé un
cancer du sein ou de l’ovaire.
La Haute Autorité de Santé a émis en 2014 des recommandations, basées sur un
classement binaire : « risque élevé de cancer » et « risque très élevé ».
L’oncogénéticien classe la famille dans l’une ou l’autre de ces catégories.
En cas de « risque très élevée » : la surveillance sénologique est
celle d’une femme mutée BRCA.
En cas de risque « élevé » :
• Avant 50 ans :
o mammographie annuelle +/- échographie
o IRM mammaire à discuter
• A partir de 50 ans :
o mammographie +/- échographie tous les 2 ans
o dans le cadre du programme national de dépistage
organisé
Certains algorithmes (Brcapro, Bodicea…) permettent de chiffrer le risque de cancer
du sein, mais ils sont cependant d’une aide limitée pour ce type de situation
(mauvaise discrimination / calibration).
TP53, Li Fraumeni et cancer du sein
La mutation du gène TP53 augmente le risque de cancer du sein de manière
majeure, quoiqu’encore imprécise.
Le cancer du sein touche alors des femmes très jeunes (âge moyen 25 ans). La
surveillance est essentiellement basée sur des IRM mammaires débutant entre 20 et
25 ans.
Il est nécessaire d’évoquer cette prédisposition devant tout cancer du sein
diagnostiqué avant l’âge de 30 ans, quelle que soit l’histoire familiale.
4

Une histoire familiale répondant aux critères de Chompret du syndrome de Li
Fraumeni, doit également orienter vers l’analyse de TP53.
Tableau 1: Critère de Chompret
I
Patient avec tumeur du spectre LFS, diagnostiquée avant l’âge de 46 ans :
• Sarcome tissus mous ou osseux
• Tumeur cérébrale
• Cancer du sein pré-ménopausique
• Corticosurrénalome
• Leucémie
• Carcinome pulmonaire bronchiolo alvéolaire
ET
Au moins un apparenté au premier ou second degré avec tumeur du spectre LFS diagnostiqué
avant 56 ans (hormis cancer du sein si cas index atteint de cancer du sein)
OU
II
Patient avec tumeurs multiples dont deux appartiennent au spectre LFS (cancers du sein multiples
exclus) ; au moins une étant diagnostiquée avant l’âge de 46 ans
OU
III
Patient avec corticosurrénalome ou tumeur des plexus choroïdes, cancer du sein avant 30 ans
quelle que soit l’histoire familiale.
La complexité de la prise en charge de ces familles Li Fraumeni résulte de
l’incertitude concernant le risque de cancers d’autres organes (sarcomes, système
nerveux central, hémopathies…), par ailleurs inaccessibles pour le moment à la
surveillance. Lorsqu’une telle prédisposition est authentifiée ou fortement suspectée,
l’indication d’analyse de TP53 et la prise en charge doivent s’effectuer dans des
centres de référence.
Enfin, la présence d’une telle mutation confère une radiosensibilité particulière,
apportant une contre-indication relative à la radiothérapie.
Autres gènes de prédisposition au cancer du sein
D’autres gènes sont impliqués de manière rarissime. STK11 (Syndrome de Peutz-
Jeghers), PTEN (maladie de Cowden) et CDH1 (prédisposition aux cancers gastriques
à cellules isolées) concernent des prédispositions dans lesquelles le cancer du sein
n’est pas au premier plan.
D’autres gènes peuvent également être impliqués, notamment RAD51C ou D, avec
une fréquence présumée très faible, ou d’autres, comme CHEK2, avec une fréquence
de mutation plus importante mais un effet très modéré en termes de risque. On peut
citer également PALB2, BARD1, ABRAXA, BRIP1… Compte-tenu des zones d’ombre,
notamment sur la pénétrance des variants identifiés, l’analyse de ces gènes ne
s’entend en 2015 que dans le cadre de projets de recherche, sans utilisation clinique
actuellement. Des études récentes concernant PALB2 laissent penser qu’à court
terme l’analyse de ce gène pourrait être inclus dans la pratique courante.
5
 6
7
6
7
1
/
7
100%